Citation: Omar Saucedo, Maia Martcheva, Abena Annor. Computing human to human Avian influenza $\mathcal{R}_0$ via transmission chains and parameter estimation[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 3465-3487. doi: 10.3934/mbe.2019174

| [1] | M. D. R. van Beest Holle and A. Meijer, Human to human transmission of avian influenza /h7n7, the netherlands, Euro Surveill., 10 (2005), 264–268. |
| [2] | K. Ungchusak, Probable person-to-person transmission of avian influenza a (h5n1), New Engl. J. Med., 352 (2005), 333–340. |
| [3] | I. Kandum, Three indonesian clusters of h5n1 virus infection in 2005, New Engl. J. Med., 355 (2006), 2186–2194. |
| [4] | S. Herfst, Airborne transmission of influenza a/h5n1 virus between ferrets, Science, 336 (2012), 1534–1541. |
| [5] | A. J. Hay, The evolution of human influenza viruses, Philos. T. R. Soc. B, 356 (2001), 1861–1870. 6. Avian influenza (2015). |
| [6] | 7. Centers for Disease Control and Prevention, Highly pathogenic avian influenza a (h5n1) virus, (2015). |
| [7] | 8. Y. Yang, Detecting human to human transmission of avian influenza a (h5n1), Emerg. Infect. Dis., 13 (2005), 1348–1353. |
| [8] | 9. J. K. Taubenberger and D. M Morens, The pathology of influenza virus infections, Annu. Rev. Pathol., 3 (2005), 499–522. |
| [9] | 10. Mathematical model, Infect. Dis., (2015). |
| [10] | 11. J. Hyman and L. Jia, An intuitive formulation for the reproductive number for the spread of disease in heterogeneous populations, Math. Biosci., 167 (2000), 65–86. |
| [11] | 12. Y. Xiao, X. Sun and S. Tang Transmission potential of the novel avian influenza a(h7n9) infection in mainland china, J. Theor. Biol., 352 (2014), 1–5. |
| [12] | 13. N. Tuncer and M. Martcheva, Modeling seasonality in avian influenza h5n1, J. Biol. Syst., 21 (2013), 1–27. |
| [13] | 14. G. Chowell, L. Simonsen, S. Towers, et al., Transmission potential of influenza h7n9 february to may 20china, BMC Medicine, 11 (2013), 1–13. |
| [14] | 15. M. van Boven, M. Koopmans, M. Du Ry van Beest Holle, et al., Detecting emerging transmissi- bility of avian influenza virus in human households, PLoS Comput. Biol., 3 (2007), 1–9. |
| [15] | 16. M. E. Woolhouse and S. Gowtage-Sequeria, Host range and emerging and reemerging pathogens, Emerg. Reemerg. Pathog., 11 (2005), 1842–1847. |
| [16] | 17. S. Blumger and J. Lloyd-Smith, Inference of R 0 and transmission heterongeneity from the size distribution of stuttering chains, PLoS Comput. Biol., 9 (2013), 1–17. |
| [17] | 18. S. Blumberg, S. Funk and J. R. Pulliam, Detecting differential transmissibilities that affect the size of self-limited outbreaks, PloS One, 10 (2014), 1–14. |
| [18] | 19. S. Blumberg and J. Lloyd-Smith, Comparing methods for estimating R 0 from the size distribution of subcritical transmission chains, Epidemics, 5 (2013), 131–145. |
| [19] | 20. V. Pitzer, Little evidence for genetic susceptibility to influenza a from family clustering data, Emerg. Infect. Dis., 13 (2007) 1074–1076. |
| [20] | 21. S. J. Olsen, Family clustering of avian influenza a (h5n1), Emerg. Infect. Dis., 11 (5), 1799– 1801. |
| [21] | 22. T. Harris, The Theory of Branching Process, Dover, 2002. |
| [22] | 23. I. Dumitriu, J. Spencer and C. Yan, Branching processes with negative offspring distribution, Ann. Comb., 7 (2003), 35–47. |
| [23] | 24. J. Lloyd-Smith, S. Schreiber, P. Kopp, et al., Superspreading and the effect of individual variation on disease emergence, Nature, 438 (2005), 355–359. |
| [24] | 25. M. Dwass, The total progeny in a branching process and a related random walk, J. Appl. Probab., 6 (1969), 682–686. |
| [25] | 26. World Health Organization, Cumulative number of confirmed human cases of avian influenza a (h5n1) reported to who (2015). |
| [26] | 27. S. Iwami, Y. Takeuchi and X. Liu, Avian flu pandemic: Can we prevent it?, J. Theor. Biol., 257 (2009), 181–190. |
| [27] | 28. W. H. O. Commmittee, The writing committee of the who consultation on human influenza a/h5 avian influenza a (h5n1) infection in humans, New Engl. J. Med., 353 (2005), 1374–1385. |
| [28] | 29. Food and Agriculture Organization of United Nations, Animal production and health division, (2015). |
| [29] | 30. Centers for Disease Control and Prevention, Avian influenza a (h7n9) virus, (2014). |
| [30] | 31. C. Hayden, Transmission of avian influenza viruses to and between humans, J. Infect. Dis., 192 (2005), 1311–1314. |
| [31] | 32. L. A. Reperant, T. Kuiken and A. D. Osterhaus, Influenza viruses, J. Hum. Vaccin. Immunother., 8 (2012), 7–16. |
| [32] | 33. A. A. King, M. Domenech de Cells, F. M. G. Magpantay, et al., Avoidable errors in modelling of outbreaks of emerging pathogens with special reference to ebola, P. Roy. Soc. B, 282 (2015), 143–151. |
| [33] | 34. M.C.Eisenberg, S.L.Robertson, J.H.Tien, Identifiabilityandestimationofmultipletransmission pathways in cholera and waterborne disease, J. Theor. Biol., 324 (2013), 84–102. |
| [34] | 35. N. Meshkat, M. Eisenberg and J. DiStefano, An algorithm for finding globally identifiable pa- rameter combinations of nonlinear ode models using grobner bases, Math. Biosci., 222 (2009), 61–72. |
| [35] | 36. M. C. Eisenberg and M. A. L. Hayashi, Determining identifiable parameter combinations using subset profiling, Math. Biosci., 256 (2014), 116–126. |
| [36] | 37. N. D. Evans, L. J. White, M. J. Chapman, et al., The structural identifiability of the susceptible infected recovered model with seasonal forcing, Math. Biosci., 194 (2005), 175–197. |
| [37] | 38. H. Kelejian, Random parameters in a simultaneous equation framework: Identification and esti- mation, Econometrica, 42 (1974), 517–527. |
| [38] | 39. H. Maio, X. Xia, A. Perelson, et al., On identifiability of nonlinear ode models and applications in viral dynamics, SIAM Rev. Soc. Ind. Appl. Math., 53 (2011), 3–39. |
| [39] | 40. N. Meshkat, C. Anderson, S. J. Rd, Alternative to ritt's pseudodivision for finding the input-output equations of multi-output models, Math. Biosci., 2(2012), 117–123. |
| [40] | 41. G. S. Bellu, M. Audoly and S. D'Angio, Daisy: A new software tool to test global identifiability of biological and physiological systems, Comput. Meth. Prog. Bio., 88 (2007), 52–61. |
| [41] | 42. A. Raue, J. Karlsson and M. Jirstrand, Comparison of approaches for parameter identifiability analysis of biological systems, Bioinformatics, 30 (2014), 1440–1448. |
| [42] | 43. H. T. Banks, J. E. Banks, C. Jackson, et al., Modeling the east coast akalat population: Model com- parison and parameter estimation, Center for Research in Scientific Computation Report, (2013), 1–39. |
| [43] | 44. A. Raue and C. Kreutz, Structural and practical identifiability analysis of partially observed dy- namical models by exploiting the profile likelihood, Oxford University Press, 25 (2009), 1923– 1929. |
| [44] | 45. N. M. Ferguson and C. Fraser, Public health risk from the avian h5n1 influenza epidemic, Science, 304 (2004), 968–969. |
| [45] | 46. Y. H. Hsieh, J. Wu, J. Fang, et al., Quantification of bird to bird and bird to human infections during 2013 novel h7n9 avian influenza outbreak in china, Lancet, 383 (2014), 541–548. |
| [46] | 47. N. Marquetoux, M. Paul, S. Wongnarkpet, et al., Estimating spatial and temporal variations of the reproductive number for highly pathogenic avian influenza h5n1 epidemic in thailand, Prev. Vet. Med., 106 (2012), 143–151. |
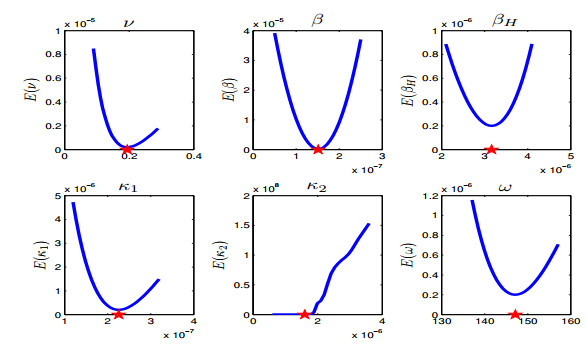
Figures(13) / Tables(5)

Omar Saucedo, Maia Martcheva, Abena Annor. Computing human to human Avian influenza $\mathcal{R}_0$ via transmission chains and parameter estimation[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 3465-3487. doi: 10.3934/mbe.2019174







 DownLoad:
DownLoad: