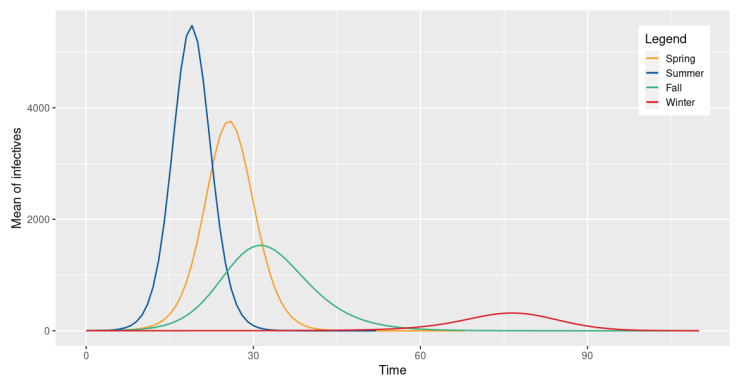
To better describe the spread of a disease, we extend a discrete time stochastic SIR-type epidemic model of Tuckwell and Williams. We assume the dependence on time of the number of daily encounters and include a parameter to represent a possible quarantine of the infectious individuals. We provide an analytic description of this Markovian model and investigate its dynamics. Both a diffusion approximation and the basic reproduction number are derived. Through several simulations, we show how the evolution of a disease is affected by the distribution of the number of daily encounters and its dependence on time. Finally, we show how the appropriate choice of this parameter allows a suitable application of our model to two real diseases.
Citation: Mireia Besalú, Giulia Binotto. Time-dependent non-homogeneous stochastic epidemic model of SIR type[J]. AIMS Mathematics, 2023, 8(10): 23218-23246. doi: 10.3934/math.20231181
To better describe the spread of a disease, we extend a discrete time stochastic SIR-type epidemic model of Tuckwell and Williams. We assume the dependence on time of the number of daily encounters and include a parameter to represent a possible quarantine of the infectious individuals. We provide an analytic description of this Markovian model and investigate its dynamics. Both a diffusion approximation and the basic reproduction number are derived. Through several simulations, we show how the evolution of a disease is affected by the distribution of the number of daily encounters and its dependence on time. Finally, we show how the appropriate choice of this parameter allows a suitable application of our model to two real diseases.

| [1] | D. Bernoulli, Essai d'une nouvelle analyse de la mortalité causée par la petite vérole, et des avantages de l'Inoculation pour la prévenir, Hist. Acad. R. Sci. Paris, 1760, 1–45. |
| [2] | N. T. Bailey, The mathematical theory of infectious diseases and its applications, London: Charles Griffin & Company Limited, 1975. |
| [3] | R. M. Anderson, R. M. May, Infectious diseases of humans. Dynamics and control, Oxford: Oxford University Press, 1991. https://doi.org/10.1002/hep.1840150131 |
| [4] | O. Diekmann, H. Heesterbeek, T. Britton, Mathematical tools for understanding infectious disease dynamics, Princeton: Princeton University Press, 2013. |
| [5] |
Y. Cai, Y. Kang, W. Wang, A stochastic SIRS epidemic model with nonlinear incidence rate, Appl. Math. Comput., 305 (2017), 221–240. https://doi.org/10.1016/j.amc.2017.02.003 doi: 10.1016/j.amc.2017.02.003

|
| [6] |
X. Bardina, M. Ferrante, C. Rovira, A stochastic epidemic model of COVID-19 disease, AIMS Math., 5 (2020), 7661–7677. https://doi.org/10.3934/math.2020490 doi: 10.3934/math.2020490
|
| [7] |
F. Flandoli, E. La Fauci, M. Riva, Individual-based Markov model of virus diffusion: Comparison with COVID-19 incubation period, serial interval and regional time series, Math. Mod. Meth. Appl. Sci., 31 (2021), 907–939. https://doi.org/10.1142/S0218202521500226 doi: 10.1142/S0218202521500226
|
| [8] |
A. McKendrick, Application of mathematics to medical problems, Proc. Edinburgh Math. Soc., 14 (1926), 98–130. https://doi.org/10.1017/S0013091500034428 doi: 10.1017/S0013091500034428
|
| [9] |
W. Kermack, A. McKendrick, A contribution to the mathematical theory of epidemics, Proc. Roy. Soc. Lond., 115 (1927), 700–721. https://doi.org/10.1098/rspa.1927.0118 doi: 10.1098/rspa.1927.0118
|
| [10] | H. Abbey, An examination of the Reed-Frost theory of epidemics, Hum. Biol., 24 (1952), 201–233. |
| [11] | L. J. S. Allen, An introduction to stochastic epidemic models, In: Mathematical Epidemiology, Berlin, Heidelberg: Springer-Verlag, 2008, 81–130. https://doi.org/10.1007/978-3-540-78911-6 |
| [12] |
H. C. Tuckwell, R. J. Williams, Some properties of a simple stochastic epidemic model of SIR type, Math. Biosci., 208 (2007), 76–97. https://doi.org/10.1016/j.mbs.2006.09.018 doi: 10.1016/j.mbs.2006.09.018
|
| [13] |
M. Ferrante, E. Ferraris, C. Rovira, On a stochastic epidemic SEIHR model and its diffusion approximation, Test, 25 (2016), 482–502. https://doi.org/10.1007/s11749-015-0465-z doi: 10.1007/s11749-015-0465-z
|
| [14] |
A. Gray, D. Greenhalgh, L. Hu, X. Mao, J. Pan, A stochastic differential equation SIS epidemic model, SIAM J. Appl. Math., 71 (2011), 876–902. https://doi.org/10.1137/10081856X doi: 10.1137/10081856X
|
| [15] |
T. Tsutsui, N. Minamib, M. Koiwai, T. Hamaokaa, I. Yamanea, K. Shimura, A stochastic-modeling evaluation of the foot-and-mouth-disease survey conducted after the outbreak in Miyazaki, Japan in 2000, Prev. Vet. Med., 61 (2003), 45–58. https://doi.org/10.1016/s0167-5877(03)00160-0 doi: 10.1016/s0167-5877(03)00160-0
|
| [16] |
S. Z. Huang, A new SEIR epidemic model with applications to the theory of eradication and control of diseases, and to the calculation of R0, Math. Biosci., 215 (2008), 84–104. https://doi.org/10.1016/j.mbs.2008.06.005 doi: 10.1016/j.mbs.2008.06.005
|
| [17] | S. E. A. Mohammed, Stochastic differential systems with memory: Theory, examples and applications, In: Stochastic Analysis and related topics Ⅵ, Boston: Birkhaüser, 1998. https://doi.org/10.1007/978-1-4612-2022-0_1 |
| [18] |
H. Wahlstrom, L. Englund, T. Carpenter, U. Emanuelson, A. Engvall, I. Vagsholm, A Reed-Frost model of the spread of tuberculosis within seven Swedish extensive farmed fallow deer herds, Prev. Vet. Med., 35 (1998), 181–193. https://doi.org/10.1016/s0167-5877(98)00061-0 doi: 10.1016/s0167-5877(98)00061-0
|
| [19] |
J. Ng, E. J. Orav, A generalized chain-binomial model with application to HIV infection, Math. Biosci., 101 (1990), 99–119. https://doi.org/10.1016/0025-5564(90)90104-7 doi: 10.1016/0025-5564(90)90104-7
|
| [20] | O. C. Ibe, Markov processes for stochastic modeling, Elsevier, 2013. |









Figures(10) / Tables(4)

Mireia Besalú, Giulia Binotto. Time-dependent non-homogeneous stochastic epidemic model of SIR type[J]. AIMS Mathematics, 2023, 8(10): 23218-23246. doi: 10.3934/math.20231181







 DownLoad:
DownLoad: