Citation: Sharmy S Mano, Koichiro Uto, Takao Aoyagi, Mitsuhiro Ebara. Fluidity of biodegradable substrate regulates carcinoma cell behavior: A novel approach to cancer therapy[J]. AIMS Materials Science, 2016, 3(1): 66-82. doi: 10.3934/matersci.2016.1.66

| [1] |
Nishimura N, Sasaki T (2008) Regulation of epithelial cell adhesion and repulsion: role of endocytic recycling. J Med Invest 55: 9–15. doi: 10.2152/jmi.55.9

|
| [2] | Sheetz MP, Felsenfeld DP, Galbraith CG (1998) Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol 8: 51–54. |
| [3] |
Koohestani F, Braundmeier AG, Mahdian A, et al. (2013) Extracellular matrix collagen alters cell proliferation and cell cycle progression of human uterine leiomyoma smooth muscle cells. PLOS ONE 8: e75844. doi: 10.1371/journal.pone.0075844
|
| [4] |
Philp D, Chen SS, Fitzgerald W, et al. (2005) Complex extracellular matrices promote tissue-specific stem cell differentiation. Stem Cells 23: 288–296. doi: 10.1634/stemcells.2002-0109
|
| [5] |
Farrelly N, Lee YJ, Oliver J, et al. (1999) Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J Cell Biol 144: 1337–1347. doi: 10.1083/jcb.144.6.1337
|
| [6] |
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406. doi: 10.1083/jcb.201102147
|
| [7] |
Pickup MW, Mouw JK, Weaver VM (2014) The extracellular matrix modulates the hallmarks of cancer. EMBO Reports 15: 1243–1253. doi: 10.15252/embr.201439246
|
| [8] | Ulrich TA, Pardo EMDJ, Kumar S (2009) The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res 69: 4167–74. |
| [9] |
Yeung T, Georges PC, Flanagan LA, et al. (2005) Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskel 60: 24–34. doi: 10.1002/cm.20041
|
| [10] |
Tilghman WR, Blais EM, Cowan CR, et al. (2012) Matrix rigidity regulates cancer cell growth by modulating cellular metabolism and protein synthesis. PLOS ONE 7: e37231. doi: 10.1371/journal.pone.0037231
|
| [11] |
Pathak A, Kumar S (2012) Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci U S A 109: 10334–10339 doi: 10.1073/pnas.1118073109
|
| [12] |
Liu J, Tan Y, Zhang H, et al. (2012) Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mat 11: 734–741. doi: 10.1038/nmat3361
|
| [13] |
Ghosh K, Ingber DE (2007) Micromechanical control of cell and tissue development: Implications for tissue engineering. Advan Drug Deliv Rev 59: 1306–1318. doi: 10.1016/j.addr.2007.08.014
|
| [14] |
Eshraghi S, Das S (2012) Micromechanical finite element modeling and experimental characterization of the compressive mechanical properties of polycaprolactone: hydroxyapatite composite scaffolds prepared by selective laser sintering for bone tissue engineering. Acta Biomater 8: 3138–3143. doi: 10.1016/j.actbio.2012.04.022
|
| [15] | Duncan R (2003) The dawning era of polymer therapeutics. Nat Rev Drug Discov 2: 347−360. |
| [16] |
Peer D, Karp JM, Hong S, et al. (2007) Nanocarriers as an emerging platform for cancer therapy. Nat Nanotech 2: 751–760. doi: 10.1038/nnano.2007.387
|
| [17] | Krishnan V, Xu X, Barwe SP, et al. (2013) Dexamethasone-loaded block copolymer nanoparticles induce leukemia cell death and enhance therapeutic efficacy: A novel application in pediatric nanomedicine. Mol Pharmaceutics 10: 2199−2210. |
| [18] |
Pathak A, Kumar S (2013) Dual anticancer drug/superparamagnetic iron oxide-loaded PLGA-based nanoparticles for cancer therapy and magnetic resonance imaging. Int J Pharm 447: 94–101. doi: 10.1016/j.ijpharm.2013.02.042
|
| [19] |
Woodruff MA, Hutmacher DW (2010) The return of a forgotten polymer: Polycaprolactone in the 21st century. Progress in Polymer Sci 35: 1217–1256. doi: 10.1016/j.progpolymsci.2010.04.002
|
| [20] | Abedalwafa M, Wang F, Wang L, et al. (2013) Biodegradable poly-epsilon-caprolactone (PCL) for tissue engineering applications: a review. Rev Adv Mater Sci 34: 123–140. |
| [21] |
Rie JV, Declercq H, Hoorick JV, et al. (2015) Cryogel-PCL combination scaffolds for bone tissue repair. J Mater Sci Mater Med 26:123. doi: 10.1007/s10856-015-5465-8
|
| [22] |
Uto K, Muroya T, Okamoto M, et al. (2012) Design of super-elastic biodegradable scaffolds with longitudinally oriented microchannels and optimization of the channel size for schwann cell migration. Sci Technol Adv Mater 13: 064207. doi: 10.1088/1468-6996/13/6/064207
|
| [23] |
Uto K, Yamamoto K, Hirase S, et al. (2006) Temperature-responsive cross-linked poly(ε-caprolactone) membrane that functions near body temperature. J Control Release 110: 408–413. doi: 10.1016/j.jconrel.2005.10.024
|
| [24] |
Ebara M, Uto K, Idota N, et al. (2012) Shape-memory surface with dynamically tunable nano-geometry activated by body heat. Adv Mater 24: 273–278. doi: 10.1002/adma.201102181
|
| [25] |
Versaevel M, Grevesse T, Gabriele S (2012) Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat Commun 3: 671. doi: 10.1038/ncomms1668
|
| [26] |
Forte G, Pagliari S, Ebara M, et al. (2012) Substrate stiffness modulates gene expression and phenotype in neonatal cardiomyocytes in vitro. Tissue Eng Part A 18: 1837–1848. doi: 10.1089/ten.tea.2011.0707
|
| [27] |
Romanazzo S, Forte G, Ebara M, et al. (2012) Substrate stiffness affects skeletal myoblast differentiation in vitro. Sci Technol Adv Mater 13: 064211. doi: 10.1088/1468-6996/13/6/064211
|
| [28] |
Uto K, Ebara M, Aoyagi T (2014) Temperature-responsive poly(ε-caprolactone) cell culture platform with dynamically tunable nano-roughness and elasticity for control of myoblast morphology. Int J Mol Sci 15: 1511–1524. doi: 10.3390/ijms15011511
|
| [29] |
Sell SA, Wolfe PS, Garg K (2010) The use of natural polymers in tissue engineering: a focus on electrospun extracellular matrix analogues. Polymers 2: 522–553. doi: 10.3390/polym2040522
|
| [30] | Gunatillake PA, Adhikari R (2003) Biodegradable synthetic polymers for tissue engineering. European Cells and Mat 5: 1–16. |
| [31] |
Breuls RGM, Jiya TU, Smit TH (2008) Scaffold stiffness influences cell behavior: opportunities for skeletal tissue engineering. Open Orthopedics 2: 103–109. doi: 10.2174/1874325000802010103
|
| [32] | Park JS, Chu JS, Tsou AD (2011) The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-b. Biomaterials 32: 3921–3930. |
| [33] | Ni Y, Chiang MYM (2007) Cell morphology and migration linked to substrate rigidity. Soft Matter 3: 1285–1292. |
| [34] | Yeung T, Georges PC, Flanagan LA (2005) Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton 60: 24–34. |
| [35] | Tilghman RW, Cowan CR, Mih JD, et al. (2010) Matrix rigidity regulates cancer cell growth and cellular phenotype. PLOS ONE 5: 9. |
| [36] |
Wozniak MA, Modzelewska K, Kwong L, et al. (2004) Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692: 103–119. doi: 10.1016/j.bbamcr.2004.04.007
|
| [37] |
Kroemer G, Galluzzi L, Vandenabeele P, et al. (2009) Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ 16: 3–11. doi: 10.1038/cdd.2008.150
|
| [38] |
Campisi J, Fagagna FDD (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740. doi: 10.1038/nrm2233
|
| [39] | Chen QM, Liu J, Merrett JB (2000) Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J 15: 543–551. |
| [40] |
Assoian RK, Klein EA (2008) Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol 18: 347–352. doi: 10.1016/j.tcb.2008.05.002
|
| [41] | Vicencio JM, Galluzzi L, Tajeddine N, et al. (2008) Senescence, apoptosis or autophagy? when a damaged cell must decide its path- A mini review. Gerontology 54: 92–99. |
| [42] |
Johnson DG, Walker CL (1999) Cyclins and cell cycle checkpoints. Annu Rev Pharm Tox 39: 295–312. doi: 10.1146/annurev.pharmtox.39.1.295
|
| [43] | Davis PK, Ho A, Dowdy SF (2001) Biological methods for cell-cycle synchronization of mammalian cells. Bio Techniques 30: 1322–1331. |
| [44] | Tian Y, Luo C, Lu Y, et al. (2012) Cell cycle synchronization by nutrient modulation, Integr Biol (Camb) 4: 328–334. |
| [45] |
Lee WC, Bhagat AAS, Huang S, et al. (2011) High-throughput cell cycle synchronization using inertial forces in spiral microchannels. Lab Chip 11: 1359–1367. doi: 10.1039/c0lc00579g
|
| [46] |
Chen M, Huang J, Yang X, et al. (2012) Serum starvation induced cell cycle synchronization facilitates human somatic cells reprogramming. PLOS ONE 7: e28203. doi: 10.1371/journal.pone.0028203
|
| [47] | Gstraunthaler G (2003) Alternatives to the use of fetal bovine serum: Serum-free cell culture, ALTEX 20: 275–281. |
| [48] |
Eric AK, Liqun Y, Devashish K, et al. (2009) Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol 19: 1511–1518. doi: 10.1016/j.cub.2009.07.069
|
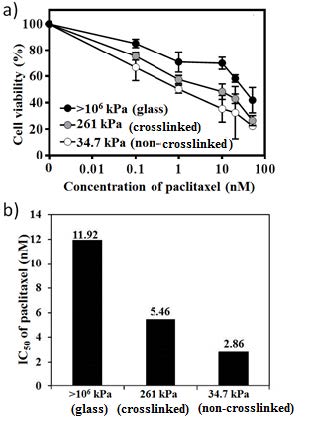
| [49] | Özdemir O (2011) Negative impact of paclitaxel crystallization on hydrogels and novel approaches for anticancer drug delivery systems, Current cancer treatment- Novel beyond conventional approaches. In Tech Open, Croatia 767–782 |
| [50] |
Chiang PC, Goul S, Nannini M (2014) Nanosuspension delivery of paclitaxel to xenograft mice can alter drug disposition and anti-tumor activity. Nanoscale Res Lett 9: 156. doi: 10.1186/1556-276X-9-156
|
| [51] |
Liebmann JE, Cook JA, Lipschultz C, et al. (1993) Cytotoxic studies of pacfitaxel (Taxol®) in human tumour cell lines. Br J Cancer 68: 1104–1109. doi: 10.1038/bjc.1993.488
|

Figures(6)

Sharmy S Mano, Koichiro Uto, Takao Aoyagi, Mitsuhiro Ebara. Fluidity of biodegradable substrate regulates carcinoma cell behavior: A novel approach to cancer therapy[J]. AIMS Materials Science, 2016, 3(1): 66-82. doi: 10.3934/matersci.2016.1.66







 DownLoad:
DownLoad: