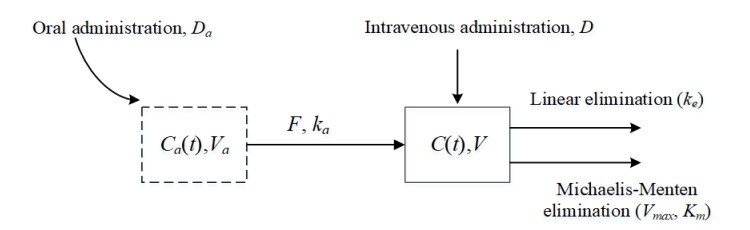
This study examined a single-compartment pharmacokinetic model with intravenous and oral administration. It investigated the trends in steady-state drug exposure and average steady-state plasma drug concentration under multiple dosing regimens. First, the combined drug administration model was a non-autonomous system, and we approximated the solution of the model by estimating the exact upper and lower bounds in conjunction with the comparison theorem for differential equations. We also demonstrated the existence, uniqueness, and stability of the solution. Second, we derived the steady-state drug exposure and compared it with the solution of the intravenous drug delivery model alone. The results indicate that the combined drug delivery scheme offers superior performance. Finally, we theoretically proved the change rule of average steady-state blood drug concentration under different dosing regimens, and verified its feasibility and rationality by combining the numerical simulation results of Phenytoin sodium.
Citation: Jiali Shi, Zhongyi Xiang. Mathematical analysis of nonlinear combination drug delivery[J]. Electronic Research Archive, 2025, 33(3): 1812-1835. doi: 10.3934/era.2025082
This study examined a single-compartment pharmacokinetic model with intravenous and oral administration. It investigated the trends in steady-state drug exposure and average steady-state plasma drug concentration under multiple dosing regimens. First, the combined drug administration model was a non-autonomous system, and we approximated the solution of the model by estimating the exact upper and lower bounds in conjunction with the comparison theorem for differential equations. We also demonstrated the existence, uniqueness, and stability of the solution. Second, we derived the steady-state drug exposure and compared it with the solution of the intravenous drug delivery model alone. The results indicate that the combined drug delivery scheme offers superior performance. Finally, we theoretically proved the change rule of average steady-state blood drug concentration under different dosing regimens, and verified its feasibility and rationality by combining the numerical simulation results of Phenytoin sodium.

| [1] |
R. Panchagnula, N. S. Thomas, Biopharmaceutics and pharmacokinetics in drug research, Int. J. Pharm., 201 (2000), 131–150. https://doi.org/10.1016/s0378-5173(00)00344-6 doi: 10.1016/s0378-5173(00)00344-6

|
| [2] | M. A. Hedaya, Basic Pharmacokinetics, Routledge, 2023. https://doi.org/10.1016/b978-0-12-815499-1.00050-8 |
| [3] | X. H. Huang, Q. S. Zheng, Pharmacokinetic and pharmacodynamic data analysis: concepts and applications, Am. J. Pharm. Educ., 74 (2010). https://doi.org/10.5688/aj740353b |
| [4] |
C. Csajka, D. Verotta, Pharmacokinetic–pharmacodynamic modelling: history and perspectives, J. Pharmacokinet. Pharmacodyn., 33 (2006), 227–279. https://doi.org/10.1007/s10928-005-9002-0 doi: 10.1007/s10928-005-9002-0
|
| [5] |
W. Wang, E. Q. Wang, J. P. Balthasar, Monoclonal antibody pharmacokinetics and pharmacodynamics, Clin. Pharmacol. Ther., 84 (2008), 548–558. https://doi.org/10.1038/clpt.2008.170 doi: 10.1038/clpt.2008.170
|
| [6] |
Y. M. C. Wang, B. Sloey, T. Wong, P. Khandelwal, R. Melara, Y. N. Sun, Investigation of the pharmacokinetics of romiplostim in rodents with a focus on the clearance mechanism, Pharm. Res., 28 (2011), 1931–1938. https://doi.org/10.1007/s11095-011-0420-y doi: 10.1007/s11095-011-0420-y
|
| [7] |
S. Kozawa, N. Yukawa, J. Liu, A. Shimamoto, E. Kakizaki, T. Fujimiya, Effect of chronic ethanol administration on disposition of ethanol and its metabolites in rat, Alcohol, 41 (2007), 87–93. https://doi.org/10.1016/j.alcohol.2007.03.002 doi: 10.1016/j.alcohol.2007.03.002
|
| [8] |
P. H. van der Graaf, N. Benson, L. A. Peletier, Topics in mathematical pharmacology, J. Dyn. Differ. Equations, 28 (2016), 1337–1356. https://doi.org/10.1007/s10884-015-9468-4 doi: 10.1007/s10884-015-9468-4
|
| [9] |
H. Moore, R. Allen, What can mathematics do for drug development?, Bull. Math. Biol., 81 (2019), 3421–3424. https://doi.org/10.1007/s11538-019-00632-x doi: 10.1007/s11538-019-00632-x
|
| [10] |
S. Tang, Y. Xiao, One-compartment model with Michaelis-Menten elimination kinetics and therapeutic window: an analytical approach, J. Pharmacokinet. Pharmacodyn., 34 (2007), 807–827. https://doi.org/10.1007/s10928-007-9070-4 doi: 10.1007/s10928-007-9070-4
|
| [11] |
A. Dokoumetzidis, R. Magin, P. Macheras, Fractional kinetics in multi-compartmental systems, J. Pharmacokinet. Pharmacodyn., 37, (2010), 507–524. https://doi.org/10.1007/s10928-010-9170-4 doi: 10.1007/s10928-010-9170-4
|
| [12] |
X. Wu, J. Li, F. Nekka, Closed form solutions and dominant elimination pathways of simultaneous first-order and Michaelis–Menten kinetics, J. Pharmacokinet. Pharmacodyn., 42 (2015), 151–161. https://doi.org/10.1007/s10928-015-9407-3 doi: 10.1007/s10928-015-9407-3
|
| [13] |
X. Wu, F. Nekka, J. Li, Analytical solution and exposure analysis of a pharmacokinetic model with simultaneous elimination pathways and endogenous production: The case of multiple dosing administration, Bull. Math. Biol., 81 (2019), 3436–3459. https://doi.org/10.1007/s11538-019-00651-8 doi: 10.1007/s11538-019-00651-8
|
| [14] |
N. A. Daryakenari, M. De Florio, K. Shukla, G. E. Karniadakis, Al-Aristotle: A physics-informed framework for systems biologygray-box identification, PLoS Comput. Biol., 20 (2024), e1011916. https://doi.org/10.1371/journal.pcbi.1011916 doi: 10.1371/journal.pcbi.1011916
|
| [15] |
N. A. Daryakenari, S. Wang, G. Karniadakis, CMINNs: Compartment model informed neural networks-Unlocking drug dynamics, Comput. Biol. Med., 184, (2025), 109392. https://doi.org/10.1016/j.compbiomed.2024.109392 doi: 10.1016/j.compbiomed.2024.109392
|
| [16] |
R. Panaccione, S. Ghosh, S. Middleton, J. R. Márquez, B. B. Scott, L. Flint, et al., Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis, Gastroenterology, 146 (2014), 392–400. https://doi.org/10.1053/j.gastro.2013.10.052 doi: 10.1053/j.gastro.2013.10.052
|
| [17] |
N. M. Maruthur, E. Tseng, S. Hutfless, L. M. Wilson, C. Suarez-Cuervo, Z. Berger, et al., Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis, Ann. Intern. Med., 164 (2016), 740–751. https://doi.org/10.7326/M15-2650 doi: 10.7326/M15-2650
|
| [18] |
Y. Ma, Q. Chen, Y. Zhang, J. Xue, Q. Liu, Y. Zhao, et al., Pharmacokinetics, safety, tolerability, and feasibility of apatinib in combination with gefitinib in stage IIIB-IV EGFR-mutated non-squamous NSCLC: a drug-drug interaction study, Cancer Chemother. Pharmacol., 92 (2023), 411–418. https://doi.org/10.1007/s00280-023-04563-2 doi: 10.1007/s00280-023-04563-2
|
| [19] |
L. Hanum, Q. Ertiningsih, N. Susyanto, Sensitivity analysis unveils the interplay of drug-sensitive and drug-resistant Glioma cells: Implications of chemotherapy and anti-angiogenic therapy, Electron. Res. Arch., 32 (2024), 72–89. https://doi.org/10.3934/era.2024004 doi: 10.3934/era.2024004
|
| [20] |
X. Tan, S. Fan, K. Duan, M. Xu, J. Zhang, P. Sun, et al., A novel drug-drug interactions prediction method based on a graph attention network, Electron. Res. Arch., 31 (2023), 5632–5648. https://doi.org/10.3934/era.2023286 doi: 10.3934/era.2023286
|
| [21] |
N. Erawaty, N. Aris, A mathematical study of effects of delays arising from the interaction of anti-drug antibody and therapeutic protein in the immune response system, AIMS Math., 5 (2020), 7176–7199. https://doi.org/10.3934/math.2020460 doi: 10.3934/math.2020460
|
| [22] |
T. Gould, R. J. Roberts, Therapeutic problems arising from the use of the intravenous route for drug administration, J. Pediatr., 95 (1979), 465–471. https://doi.org/10.1016/s0022-3476(79)80538-7 doi: 10.1016/s0022-3476(79)80538-7
|
| [23] |
P. Fasinu, V. Pillay, V. M. K. Ndesendo, L. C. du Toit, Y. E. Choonara, Diverse approaches for the enhancement of oral drug bioavailability, Biopharm. Drug Dispos., 32 (2011), 185–209. https://doi.org/10.1002/bdd.750 doi: 10.1002/bdd.750
|
| [24] |
R. Selimov, E. Goncharova, P. Koriakovtsev, D. Gabidullina, J. Karsakova, S. Kozlov, et al., Comparative pharmacokinetics of florfenicol in heifers after intramuscular and subcutaneous administration, J. Vet. Pharmacol. Ther., 46 (2023), 177–184. https://doi.org/10.1111/jvp.13110 doi: 10.1111/jvp.13110
|
| [25] |
N. Z. Kerin, R. D. Blevins, H. Frumin, K. Faitel, M. Rubenfire, Intravenous and oral loading versus oral loading alone with amiodarone for chronic refractory ventricular arrhythmias, Am. J. Cardiol., 55 (1985), 89–91. https://doi.org/10.1016/0002-9149(85)90305-4 doi: 10.1016/0002-9149(85)90305-4
|
| [26] |
H. U. Xiao-hu, T. San-yi, Approximate Solutions to the Nonlinear Compartmental Model for Extravascular Administration, Appl. Math. Mech., 35 (2014), 1033–1045. https://doi.org/10.3879/j.issn.1000-0887.2014.09.009 doi: 10.3879/j.issn.1000-0887.2014.09.009
|
| [27] |
S. H. Jang, P. M. Colangelo, J. V. S. Gobburu, Exposure-response of posaconazole used for prophylaxis against invasive fungal infections: evaluating the need to adjust doses based on drug concentrations in plasma, Clin. Pharmacol. Ther., 88 (2010), 115–119. https://doi.org/10.1038/clpt.2010.64 doi: 10.1038/clpt.2010.64
|
| [28] | M. Prasad, P. R. Krishnan, R. Sequeira, Khaldoon. Al-Roomi, Anticonvulsant therapy for status epilepticus, Cochrane Database Syst. Rev., 2014. https://doi.org/10.1002/14651858.CD003723.pub3 |
| [29] |
E. H. Reynolds, D. Chadwick, A. W. Galbraith, One drug(phenytoin) in the treatment of epilepsy, The lancet, 307 (1976), 923–926. https://doi.org/10.1016/s0140-6736(76)92881-6 doi: 10.1016/s0140-6736(76)92881-6
|
| [30] |
J. Patocka, Q. Wu, E. Nepovimova, K. Kuca, Phenytoin–An anti-seizure drug: Overview of its chemistry, pharmacology and toxicology, Food Chem. Toxicol., 142 (2020), 111393. https://doi.org/10.1016/j.fct.2020.111393 doi: 10.1016/j.fct.2020.111393
|
| [31] |
W. J. Jusko, J. R. Koup, G. Alván, Nonlinear assessment of phenytoin bioavailability, J. Pharmacokinet. Biopharm., 4 (1976), 327–336. https://doi.org/10.1007/bf01063122 doi: 10.1007/bf01063122
|
| [32] |
R. Gugler, C. V. Manion, D. L. Azarnoff, Phenytoin: pharmacokinetics and bioavailability, Clin. Pharmacol. Ther., 19 (1976), 135–142. https://doi.org/10.1002/cpt1976192135 doi: 10.1002/cpt1976192135
|
| [33] |
L. Osterberg, T. Blaschke, Adherence to medication, N. Engl. J. Med., 353 (2005), 487–497. https://doi.org/10.7748/ns.25.2.59.s53 doi: 10.7748/ns.25.2.59.s53
|
| [34] |
B. Vrijens, S. De Geest, D. A. Hughes, K. Przemyslaw, J. Demonceau, T. Ruppar, et al., A new taxonomy for describing and defining adherence to medications, Br. J. Clin. Pharmacol., 73 (2012), 691–705. https://doi.org/10.1111/j.1365-2125.2012.04167.x doi: 10.1111/j.1365-2125.2012.04167.x
|
| [35] |
M. E. Csete, J. C. Doyle, Reverse engineering of biological complexity, Science, 295 (2002), 1664–1669. https://doi.org/10.1126/science.1069981 doi: 10.1126/science.1069981
|
| [36] |
B. Munos, Lessons from 60 years of pharmaceutical innovation, Nat. Rev. Drug Discov., 8 (2009), 959–968. https://doi.org/10.1038/nrd2961 doi: 10.1038/nrd2961
|






Figures(7)
Jiali Shi, Zhongyi Xiang. Mathematical analysis of nonlinear combination drug delivery[J]. Electronic Research Archive, 2025, 33(3): 1812-1835. doi: 10.3934/era.2025082







 DownLoad:
DownLoad: