Seven tesla magnetic resonance imaging (7T MRI) is known to offer a superior spatial resolution and a signal-to-noise ratio relative to any other non-invasive imaging technique and provides the possibility for neuroimaging researchers to observe disease-related structural changes, which were previously only apparent on post-mortem tissue analyses. Alzheimer's disease is a natural and widely used subject for this technology since the 7T MRI allows for the anticipation of disease progression, the evaluation of secondary prevention measures thought to modify the disease trajectory, and the identification of surrogate markers for treatment outcome. In this editorial, we discuss the various neuroimaging biomarkers for Alzheimer's disease that have been studied using 7T MRI, which include morphological alterations, molecular characterization of cerebral T2*-weighted hypointensities, the evaluation of cerebral microbleeds and microinfarcts, biochemical changes studied with MR spectroscopy, as well as some other approaches. Finally, we discuss the limitations of the 7T MRI regarding imaging Alzheimer's disease and we provide our outlook for the future.
Citation: Arosh S. Perera Molligoda Arachchige, Anton Kristoffer Garner. Seven Tesla MRI in Alzheimer's disease research: State of the art and future directions: A narrative review[J]. AIMS Neuroscience, 2023, 10(4): 401-422. doi: 10.3934/Neuroscience.2023030
Seven tesla magnetic resonance imaging (7T MRI) is known to offer a superior spatial resolution and a signal-to-noise ratio relative to any other non-invasive imaging technique and provides the possibility for neuroimaging researchers to observe disease-related structural changes, which were previously only apparent on post-mortem tissue analyses. Alzheimer's disease is a natural and widely used subject for this technology since the 7T MRI allows for the anticipation of disease progression, the evaluation of secondary prevention measures thought to modify the disease trajectory, and the identification of surrogate markers for treatment outcome. In this editorial, we discuss the various neuroimaging biomarkers for Alzheimer's disease that have been studied using 7T MRI, which include morphological alterations, molecular characterization of cerebral T2*-weighted hypointensities, the evaluation of cerebral microbleeds and microinfarcts, biochemical changes studied with MR spectroscopy, as well as some other approaches. Finally, we discuss the limitations of the 7T MRI regarding imaging Alzheimer's disease and we provide our outlook for the future.
beta-amyloid
Alzheimer's disease
Arterial Spin Labelling
blood oxygenation level-dependent
Cornus Ammonis
stratum radiatum and lacunosum-moleculare of the Cornu Ammonis' field 1
cerebral blood flow
chemical exchange saturation transfer
cerebral microinfarcts
contrast-to-noise ratio
cerebrospinal fluid
diffusion kurtosis imaging
diffusion tensor imaging
electroencephalography
fluid attenuated inversion recovery
functional MRI
glutamine
glutamate
Chemical Exchange Saturation Transfer of glutamate
γ-Aminobutyric acid
gradient recalled echo
locus coeruleus
mild cognitive impairment
myo-inositol
magnetization transfer
MR Spectroscopy
Positron Emission Tomography
perivascular spaces
Pittsburgh compound B
radiofrequency
signal-to-noise ratio
susceptibility-weighted imaging
turbo spin echo
ultra-high field

| [1] |
Masters CL, Bateman R, Blennow K, et al. (2015) Alzheimer's disease. Nat Rev Dis Primers 1. https://doi.org/10.1038/nrdp.2015.56 
|
| [2] |
Prince M, Bryce R, Albanese E, et al. (2013) The global prevalence of dementia: a systematic review and meta-analysis. Alzheimers Dement 9: 63-75.e2. https://doi.org/10.1016/j.jalz.2012.11.007
|
| [3] |
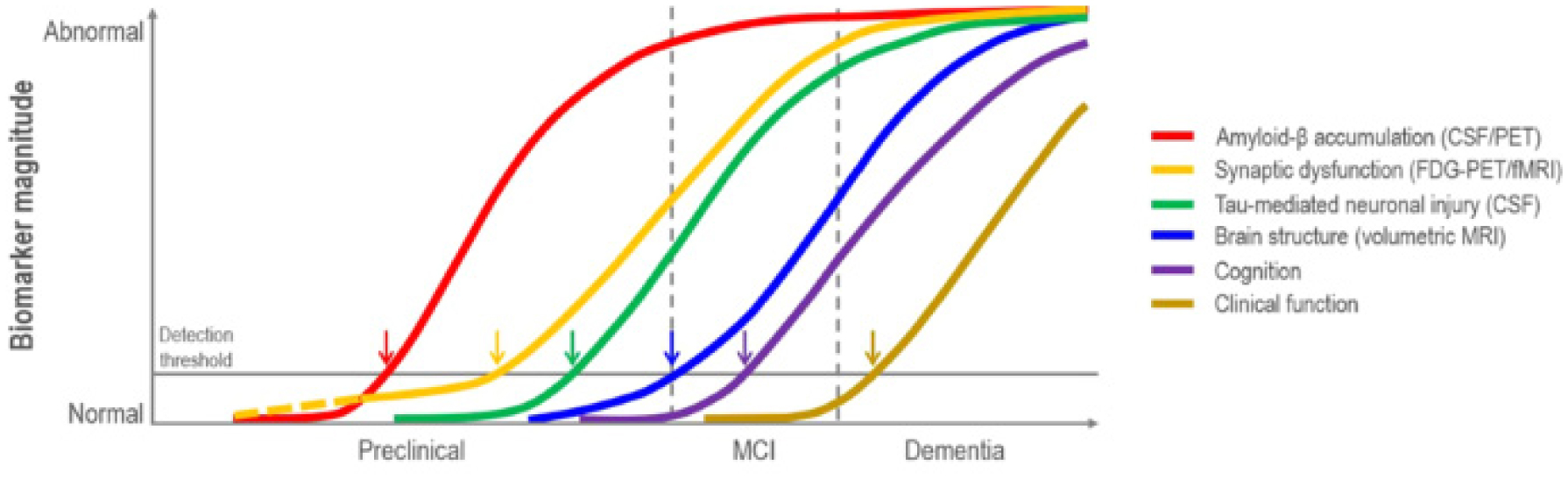
Jack CR, Knopman DS, Jagust WJ, et al. (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9: 119-128. https://doi.org/10.1016/S1474-4422(09)70299-6
|
| [4] |
Lee BC, Mintun M, Buckner RL, et al. (2003) Imaging of Alzheimer's disease. J Neuroimaging 13: 199-214. https://doi.org/10.1111/j.1552-6569.2003.tb00179.x
|
| [5] |
McKhann GM, Knopman DS, Chertkow H, et al. (2011) The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7: 263-269. https://doi.org/10.1016/j.jalz.2011.03.005
|
| [6] |
Knopman DS, Amieva H, Petersen RC, et al. (2021) Alzheimer disease. Nat Rev Dis Primers 7: 33. https://doi.org/10.1038/s41572-021-00269-y
|
| [7] |
Arachchige ASPM (2022) 7-Tesla PET/MRI: A promising tool for multimodal brain imaging?. AIMS Neurosci 9: 516-518. https://doi.org/10.3934/Neuroscience.2022029
|
| [8] |
Okada T, Fujimoto K, Fushimi Y, et al. (2022) Neuroimaging at 7 Tesla: a pictorial narrative review. Quant Imag Med Surg 12: 3406-3435. https://doi.org/10.21037/qims-21-969
|
| [9] |
Zwanenburg JJ, van der Kolk AG, Luijten PR (2013) Ultra-High-Field MR Imaging: Research Tool or Clinical Need?. PET Clin 8: 311-328. https://doi.org/10.1016/j.cpet.2013.03.004
|
| [10] |
Apostolova LG, Zarow C, Biado K (2015) Relationship between hippocampal atrophy and neuropathology markers: a 7T MRI validation study of the EADC-ADNI Harmonized Hippocampal Segmentation Protocol. Alzheimers Dement 11: 139-150. https://doi.org/10.1016/j.jalz.2015.01.00
|
| [11] |
Düzel E, Costagli M, Donatelli G, et al. (2021) Studying Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis with 7-T magnetic resonance. Eur Radiol Exp 5. https://doi.org/10.1186/s41747-021-00221-5
|
| [12] |
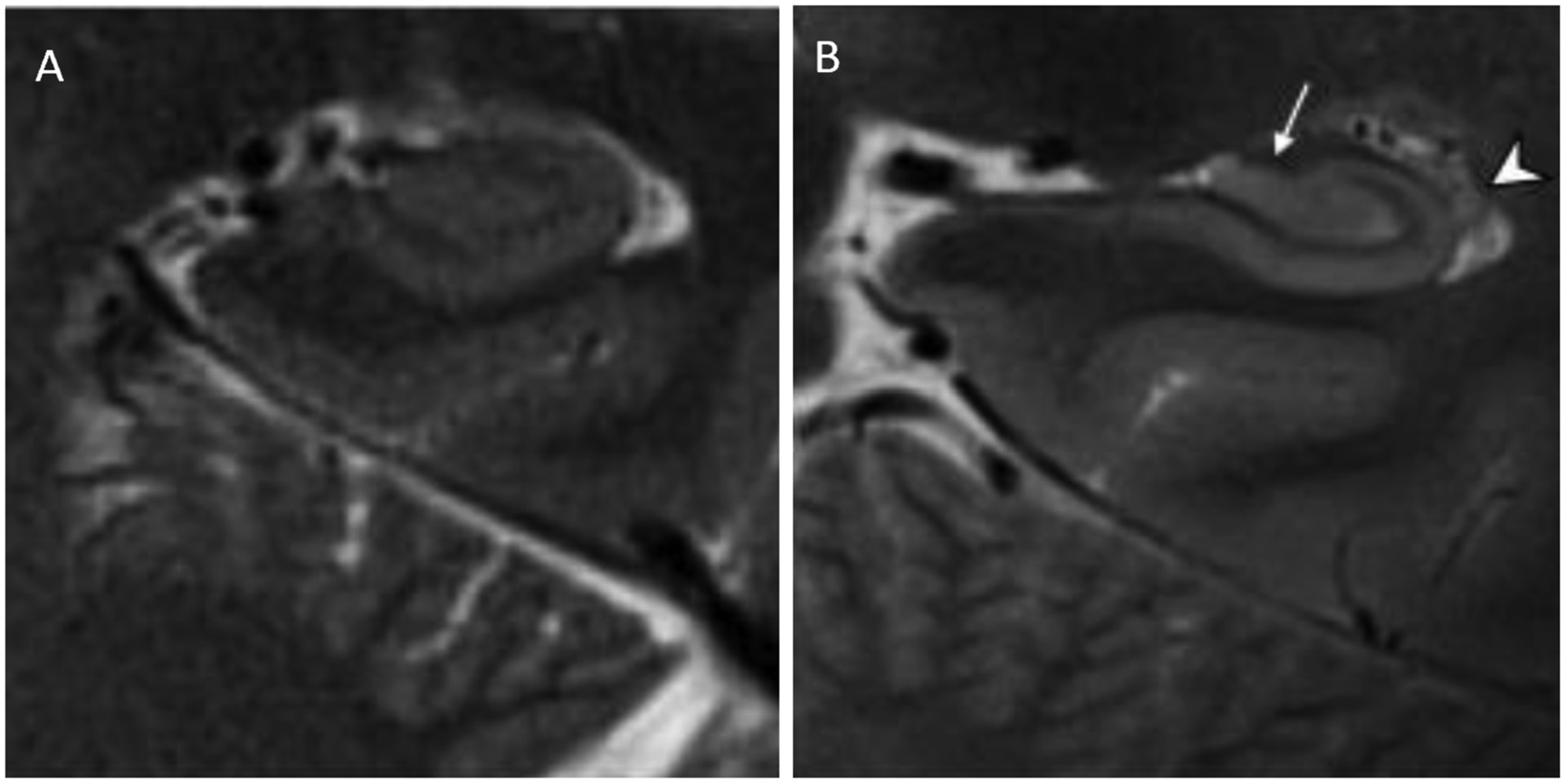
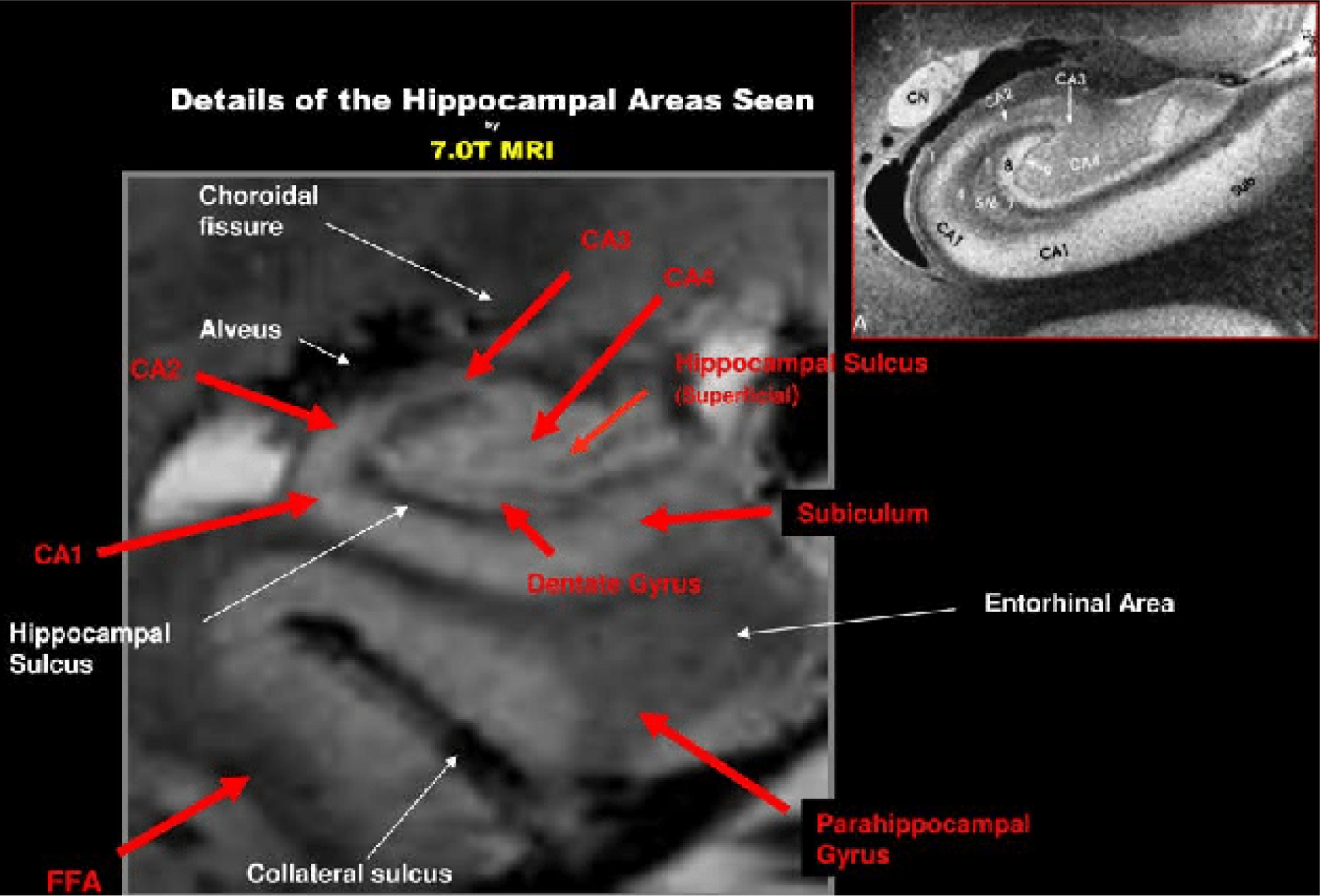
Theysohn JM, Kraff O, Maderwald S, et al. (2009) The human hippocampus at 7 T--in vivo MRI. Hippocampus 19: 1-7. https://doi.org/10.1002/hipo.20487
|
| [13] |
Kerchner GA, Hess CP, Hammond-Rosenbluth KE, et al. (2010) Hippocampal CA1 apical neuropil atrophy in mild Alzheimer disease visualized with 7-T MRI. Neurology 75: 1381-1387. https://doi.org/10.1212/WNL.0b013e3181f736a1
|
| [14] |
Barazany D, Assaf Y (2012) Visualization of cortical lamination patterns with magnetic resonance imaging. Cereb Cortex 22: 2016-2023. https://doi.org/10.1093/cercor/bhr277
|
| [15] |
Deng M, Yu R, Wang L, et al. (2016) Learning-based 3T brain MRI segmentation with guidance from 7T MRI labeling. Med Phys 43. https://doi.org/10.1118/1.4967487
|
| [16] | Cheng PW, Chiueh TD, Chen JH (2021) A high temporal/spatial resolution neuro-architecture study of rodent brain by wideband echo planar imaging. Sci Rep 11. https://doi.org/10.1038/s41598-021-98132-3 |
| [17] |
Yoo PE, John SE, Farquharson S, et al. (2018) 7T-fMRI: Faster temporal resolution yields optimal BOLD sensitivity for functional network imaging specifically at high spatial resolution. NeuroImage 164: 214-229. https://doi.org/10.1016/j.neuroimage.2017.03.002
|
| [18] | Das N, Ren J, Spence J, et al. (2021) Phosphate Brain Energy Metabolism and Cognition in Alzheimer's Disease: A Spectroscopy Study Using Whole-Brain Volume-Coil 31Phosphorus Magnetic Resonance Spectroscopy at 7Tesla. Front Neurosci 15. https://doi.org/10.3389/fnins.2021.641739 |
| [19] |
Dusek P, Dezortova M, Wuerfel J (2013) Imaging of Iron. Int Rev Neurobiol 110: 195-239. https://doi.org/10.1016/b978-0-12-410502-7.00010-7
|
| [20] |
Kim S, Lee Y, Jeon CY, et al. (2020) Quantitative magnetic susceptibility assessed by 7T magnetic resonance imaging in Alzheimer's disease caused by streptozotocin administration. Quant Imag Med Surg 10: 789-797. https://doi.org/10.21037/qims.2020.02.08
|
| [21] |
Pohmann R, Speck O, Scheffler K (2016) Signal-to-noise ratio and MR tissue parameters in human brain imaging at 3, 7, and 9.4 tesla using current receive coil arrays. Magn Reson Med 75: 801-809. https://doi.org/10.1002/mrm.25677
|
| [22] |
Uludağ K, Müller-Bierl B, Uğurbil K (2009) An integrative model for neuronal activity-induced signal changes for gradient and spin echo functional imaging. Neuroimage 48: 150-165. https://doi.org/10.1016/j.neuroimage.2009.05.051
|
| [23] |
Hazra A, Gu F, Aulakh A, et al. (2013) Inhibitory neuron and hippocampal circuit dysfunction in an aged mouse model of Alzheimer's disease. PloS One 8: e64318. https://doi.org/10.1371/journal.pone.0064318
|
| [24] | Chen C, Ma X, Wei J, et al. (2022) Early impairment of cortical circuit plasticity and connectivity in the 5XFAD Alzheimer's disease mouse model. Transl Psychiat 12. https://doi.org/10.1038/s41398-022-02132-4 |
| [25] |
Yang J, Huber L, Yu Y, et al. (2021) Linking cortical circuit models to human cognition with laminar fMRI. Neurosci Biobehav R 128: 467-478. https://doi.org/10.1016/j.neubiorev.2021.07.005
|
| [26] |
Korte N, Nortley R, Attwell D (2020) Cerebral blood flow decrease as an early pathological mechanism in Alzheimer's disease. Acta Neuropathol 140: 793-810. https://doi.org/10.1007/s00401-020-02215-w
|
| [27] |
Kashyap S, Ivanov D, Havlicek M, et al. (2021) Sub-millimetre resolution laminar fMRI using Arterial Spin Labelling in humans at 7 T. PloS One 16: e0250504. https://doi.org/10.1371/journal.pone.0250504
|
| [28] |
Shao X, Guo F, Shou Q, et al. (2021) Laminar perfusion imaging with zoomed arterial spin labeling at 7 Tesla. NeuroImage 245: 118724. https://doi.org/10.1016/j.neuroimage.2021.118724
|
| [29] |
Rutland JW, Delman BN, Gill CM, et al. (2020) Emerging Use of Ultra-High-Field 7T MRI in the Study of Intracranial Vascularity: State of the Field and Future Directions. Am J Neuroradiol 41: 2-9. https://doi.org/10.3174/ajnr.A6344
|
| [30] |
Zong F, Du J, Deng X, et al. (2021) Fast Diffusion Kurtosis Mapping of Human Brain at 7 Tesla With Hybrid Principal Component Analyses. IEEE Access 9: 107965-107975. https://doi.org/10.1109/ACCESS.2021.3100546
|
| [31] |
Chu X, Wu P, Yan H, et al. (2022) Comparison of brain microstructure alterations on diffusion kurtosis imaging among Alzheimer's disease, mild cognitive impairment, and cognitively normal individuals. Front Aging Neurosci 14: 919143. https://doi.org/10.3389/fnagi.2022.919143
|
| [32] |
McKiernan EF, O'Brien JT (2017) 7T MRI for neurodegenerative dementias in vivo: a systematic review of the literature. J Neurol Neurosur Ps 88: 564-574. https://doi.org/10.1136/jnnp-2016-315022
|
| [33] |
Giuliano A, Donatelli G, Cosottini M, et al. (2017) Hippocampal subfields at ultra high field MRI: An overview of segmentation and measurement methods. Hippocampus 27: 481-494. https://doi.org/10.1002/hipo.22717
|
| [34] |
Kenkhuis B, Jonkman LE, Bulk M, et al. (2019) 7T MRI allows detection of disturbed cortical lamination of the medial temporal lobe in patients with Alzheimer's disease. NeuroImage Clin 21: 101665. https://doi.org/10.1016/j.nicl.2019.101665
|
| [35] |
Ferreira D, Pereira JB, Volpe G, et al. (2019) Subtypes of Alzheimer's Disease Display Distinct Network Abnormalities Extending Beyond Their Pattern of Brain Atrophy. Front Neuro 10: 524. https://doi.org/10.3389/fneur.2019.00524
|
| [36] |
Boutet C, Chupin M, Lehéricy S, et al. (2014) Detection of volume loss in hippocampal layers in Alzheimer's disease using 7 T MRI: a feasibility study. NeuroImage Clin 5: 341-348. https://doi.org/10.1016/j.nicl.2014.07.011
|
| [37] |
Balchandani P, Naidich TP (2015) Ultra-High-Field MR Neuroimaging. Am J Neuroradiol 36: 1204-1215. https://doi.org/10.3174/ajnr.A4180
|
| [38] |
Riphagen JM, Schmiedek L, Gronenschild EHBM, et al. (2020) Associations between pattern separation and hippocampal subfield structure and function vary along the lifespan: A 7 T imaging study. Sci Rep 10: 7572. https://doi.org/10.1038/s41598-020-64595-z
|
| [39] |
Kamsu JM, Constans JM, Lamberton F, et al. (2013) Structural layers of ex vivo rat hippocampus at 7T MRI. PloS One 8: e76135. https://doi.org/10.1371/journal.pone.0076135
|
| [40] |
Yin JX, Turner GH, Lin HJ, et al. (2011) Deficits in spatial learning and memory is associated with hippocampal volume loss in aged apolipoprotein E4 mice. J Alzheimers Dis 27: 89-98. https://doi.org/10.3233/JAD-2011-110479
|
| [41] |
Zhou X, Huang J, Pan S, et al. (2016) Neurodegeneration-Like Pathological and Behavioral Changes in an AAV9-Mediated p25 Overexpression Mouse Model. J Alzheimers Dis 53: 843-855. https://doi.org/10.3233/JAD-160191
|
| [42] |
Wisse LEM, Biessels GJ, Heringa SM, et al. (2014) Hippocampal subfield volumes at 7T in early Alzheimer's disease and normal aging. Neurobiol Aging 35: 2039-2045. https://doi.org/10.1016/j.neurobiolaging.2014.02.021
|
| [43] |
Blom K, Koek HL, Zwartbol MHT, et al. (2020) Vascular Risk Factors of Hippocampal Subfield Volumes in Persons without Dementia: The Medea 7T Study. J Alzheimers Dis 77: 1223-1239. https://doi.org/10.3233/JAD-200159
|
| [44] | Chen Y, Chen T, Hou R (2022) Locus coeruleus in the pathogenesis of Alzheimer's disease: A systematic review. Alzh Dement 8: e12257. https://doi.org/10.1002/trc2.12257 |
| [45] |
Priovoulos N, Jacobs HIL, Ivanov D, et al. (2018) High-resolution in vivo imaging of human locus coeruleus by magnetization transfer MRI at 3T and 7T. NeuroImage 168: 427-436. https://doi.org/10.1016/j.neuroimage.2017.07.045
|
| [46] |
Tuzzi E, Balla DZ, Loureiro JRA, et al. (2020) Ultra-High Field MRI in Alzheimer's Disease: Effective Transverse Relaxation Rate and Quantitative Susceptibility Mapping of Human Brain In Vivo and Ex Vivo compared to Histology. J Alzheimers Dis 73: 1481-1499. https://doi.org/10.3233/JAD-190424
|
| [47] |
van Rooden S, Versluis MJ, Liem MK, et al. (2014) Cortical phase changes in Alzheimer's disease at 7T MRI: a novel imaging marker. Alzheimers Dement 10: e19-e26. https://doi.org/10.1016/j.jalz.2013.02.002
|
| [48] |
Cai K, Tain R, Das S, et al. (2015) The feasibility of quantitative MRI of perivascular spaces at 7T. J Neurosci Meth 256: 151-156. https://doi.org/10.1016/j.jneumeth.2015.09.001
|
| [49] |
van Rooden S, Doan NT, Versluis MJ, et al. (2015) 7T T2*-weighted magnetic resonance imaging reveals cortical phase differences between early- and late-onset Alzheimer's disease. Neurobiol Aging 36: 20-26. https://doi.org/10.1016/j.neurobiolaging.2014.07.006
|
| [50] |
van Rooden S, Buijs M, van Vliet ME, et al. (2016) Cortical phase changes measured using 7-T MRI in subjects with subjective cognitive impairment, and their association with cognitive function. NMR Biomed 29: 1289-1294. https://doi.org/10.1002/nbm.3248
|
| [51] |
van Bergen JM, Li X, Hua J, et al. (2016) Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci Rep 6: 35514. https://doi.org/10.1038/srep35514
|
| [52] |
Nakada T, Matsuzawa H, Igarashi H, et al. (2008) In vivo visualization of senile-plaque-like pathology in Alzheimer's disease patients by MR microscopy on a 7T system. J Neuroimaging 18: 125-129. https://doi.org/10.1111/j.1552-6569.2007.00179.x
|
| [53] |
Bulk M, Abdelmoula WM, Nabuurs RJA, et al. (2018) Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer's disease. Neurobiol Aging 62: 231-242. https://doi.org/10.1016/j.neurobiolaging.2017.10.017
|
| [54] |
Zeineh MM, Chen Y, Kitzler HH, et al. (2015) Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging 36: 2483-2500. https://doi.org/10.1016/j.neurobiolaging.2015.05.022
|
| [55] |
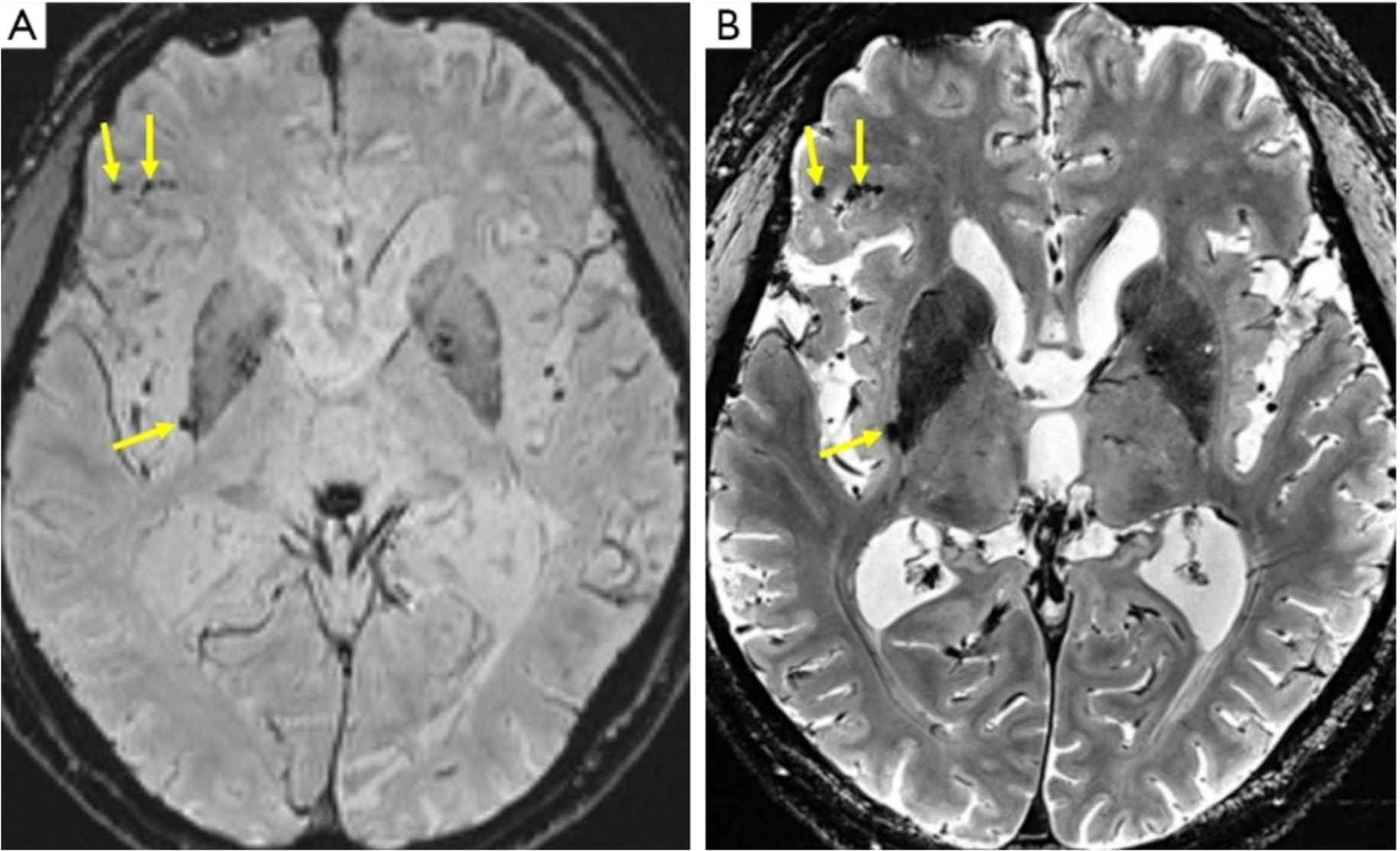
van Rooden S, Goos JD, van Opstal AM, et al. (2014) Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 270: 205-211. https://doi.org/10.1148/radiol.13130743
|
| [56] |
Rowe CC, Villemagne VL (2011) Brain amyloid imaging. J Nucl Med 52: 1733-1740. https://doi.org/10.2967/jnumed.110.076315
|
| [57] |
Schreiner SJ, Liu X, Gietl AF, et al. (2014) Regional Fluid-Attenuated Inversion Recovery (FLAIR) at 7 Tesla correlates with amyloid beta in hippocampus and brainstem of cognitively normal elderly subjects. Front Aging Neurosci 6: 240. https://doi.org/10.3389/fnagi.2014.00240
|
| [58] |
van Veluw SJ, Zwanenburg JJ, Rozemuller AJ, et al. (2015) The spectrum of MR detectable cortical microinfarcts: a classification study with 7-tesla postmortem MRI and histopathology. J Cerebr Blood F Met 35: 676-683. https://doi.org/10.1038/jcbfm.2014.258
|
| [59] |
Conijn MM, Geerlings MI, Biessels GJ, et al. (2011) Cerebral microbleeds on MR imaging: comparison between 1.5 and 7T. Am J Neuroradiol 32: 1043-1049. https://doi.org/10.3174/ajnr.A2450
|
| [60] |
Brundel M, Heringa SM, de Bresser J, et al. (2012) High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer's disease. J Alzheimers Dis 31: 259-263. https://doi.org/10.3233/JAD-2012-120364
|
| [61] |
van Veluw SJ, Biessels GJ, Klijn CJ, et al. (2016) Heterogeneous histopathology of cortical microbleeds in cerebral amyloid angiopathy. Neurology 86: 867-871. https://doi.org/10.1212/WNL.0000000000002419
|
| [62] |
Ni J, Auriel E, Martinez-Ramirez S, et al. (2015) Cortical localization of microbleeds in cerebral amyloid angiopathy: an ultra high-field 7T MRI study. J Alzheimers Dis 43: 1325-1330. https://doi.org/10.3233/JAD-140864
|
| [63] | Hütter BO, Altmeppen J, Kraff O, et al. (2020) Higher sensitivity for traumatic cerebral microbleeds at 7 T ultra-high field MRI: is it clinically significant for the acute state of the patients and later quality of life?. Ther Adv Neurol Diso 13. https://doi.org/10.1177/1756286420911295 |
| [64] |
Uğurbil K, Adriany G, Andersen P, et al. (2003) Ultrahigh field magnetic resonance imaging and spectroscopy. Magn Reson Imaging 21: 1263-1281. https://doi.org/10.1016/j.mri.2003.08.027
|
| [65] |
Terpstra M, Cheong I, Lyu T, et al. (2016) Test-retest reproducibility of neurochemical profiles with short-echo, single-voxel MR spectroscopy at 3T and 7T. Magn Reson Med 76: 1083-91. https://doi.org/10.1002/mrm.26022
|
| [66] |
McKay J, Tkáč I (2016) Quantitative in vivo neurochemical profiling in humans: where are we now?. Int J Epidemiol 45: 1339-50. https://doi.org/10.1093/ije/dyw235
|
| [67] |
Qin L, Gao J (2021) New avenues for functional neuroimaging: ultra-high field MRI and OPM-MEG. Psychoradiology 1: 165-171. https://doi.org/10.1093/psyrad/kkab014
|
| [68] |
Haeger A, Bottlaender M, Lagarde J, et al. (2021) What can 7T sodium MRI tell us about cellular energy depletion and neurotransmission in Alzheimer's disease?. Alzheimers 17: 1843-1854. https://doi.org/10.1002/alz.12501
|
| [69] |
Oeltzschner G, Wijtenburg SA, Mikkelsen M, et al. (2019) Neurometabolites and associations with cognitive deficits in mild cognitive impairment: a magnetic resonance spectroscopy study at 7 Tesla. Neurobiol Aging 73: 211-218. https://doi.org/10.1016/j.neurobiolaging.2018.09.027
|
| [70] |
Quevenco FC, Schreiner SJ, Preti MG, et al. (2019) GABA and glutamate moderate beta-amyloid related functional connectivity in cognitively unimpaired old-aged adults. NeuroImage Clin 22: 101776. https://doi.org/10.1016/j.nicl.2019.101776
|
| [71] |
Henning A, Fuchs A, Murdoch JB (2009) Slice-selective FID acquisition, localized by outer volume suppression (FIDLOVS) for (1)H-MRSI of the human brain at 7 T with minimal signal loss. NMR Biomed 22: 683-696. https://doi.org/10.1002/nbm.1366
|
| [72] |
Haris M, Nath K, Cai K, et al. (2013) Imaging of glutamate neurotransmitter alterations in Alzheimer's disease. NMR Biomed 26: 386-391. https://doi.org/10.1002/nbm.2875
|
| [73] | Cember ATJ, Nanga RPR, Reddy R (2022) Glutamate-weighted CEST (gluCEST) imaging for mapping neurometabolism: An update on the state of the art and emerging findings from in vivo applications. NMR Biomed e4780. [Advance online publication]. https://doi.org/10.1002/nbm.4780 |
| [74] |
Crescenzi R, DeBrosse C, Nanga RP, et al. (2017) Longitudinal imaging reveals subhippocampal dynamics in glutamate levels associated with histopathologic events in a mouse model of tauopathy and healthy mice. Hippocampus 27: 285-302. https://doi.org/10.1002/hipo.22693
|
| [75] |
Crescenzi R, DeBrosse C, Nanga RP, et al. (2014) In vivo measurement of glutamate loss is associated with synapse loss in a mouse model of tauopathy. NeuroImage 101: 185-192. https://doi.org/10.1016/j.neuroimage.2014.06.067
|
| [76] |
Haris M, Cai K, Singh A, et al. (2011) In vivo mapping of brain myo-inositol. NeuroImage 54: 2079-2085. https://doi.org/10.1016/j.neuroimage.2010.10.017
|
| [77] |
Marjańska M, McCarten JR, Hodges JS, et al. (2019) Distinctive Neurochemistry in Alzheimer's Disease via 7 T In Vivo Magnetic Resonance Spectroscopy. J Alzheimers Dis 68: 559-569. https://doi.org/10.3233/JAD-180861
|
| [78] |
Zenaro E, Piacentino G, Constantin G (2017) The blood-brain barrier in Alzheimer's disease. Neurobiol Dis 107: 41-56. https://doi.org/10.1016/j.nbd.2016.07.007
|
| [79] |
Rezai-Zadeh K, Gate D, Szekely CA, et al. (2009) Can peripheral leukocytes be used as Alzheimer's disease biomarkers?. Expert Rev Neurother 9: 1623-1633. https://doi.org/10.1586/ern.09.118
|
| [80] |
Robbins M, Clayton E, Kaminski Schierle GS (2021) Synaptic tau: A pathological or physiological phenomenon?. Acta Neuropathol Commun 9: 149. https://doi.org/10.1186/s40478-021-01246-y
|
| [81] |
Šišková Z, Justus D, Kaneko H, et al. (2014) Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of alzheimer's disease. Neuron 84: 1023-1033. https://doi.org/10.1016/j.neuron.2014.10.024
|
| [82] |
Palop JJ, Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17: 777-792. https://doi.org/10.1038/nrn.2016.141
|
| [83] |
Shaaban CE, Aizenstein HJ, Jorgensen DR, et al. (2017) In Vivo Imaging of Venous Side Cerebral Small-Vessel Disease in Older Adults: An MRI Method at 7T. Am J Neuroradiol 38: 1923-1928. https://doi.org/10.3174/ajnr.A5327
|
| [84] |
Poduslo JF, Wengenack TM, Curran GL, et al. (2002) Molecular targeting of Alzheimer's amyloid plaques for contrast-enhanced magnetic resonance imaging. Neurobiol Dis 11: 315-329. https://doi.org/10.1006/nbdi.2002.0550
|
| [85] |
Arachchige ASPM (2023) Transitioning from PET/MR to trimodal neuroimaging: why not cover the temporal dimension with EEG?. AIMS Neurosci 10: 1-4. https://doi.org/10.3934/Neuroscience.2023001
|
| [86] |
Duyn JH (2012) The future of ultra-high field MRI and fMRI for study of the human brain. NeuroImage 62: 1241-1248. https://doi.org/10.1016/j.neuroimage.2011.10.065
|
| [87] |
Biagi L, Cosottini M, Tosetti M (2017) 7 T MR: from basic research to human applications. High Field Brain MRI . Cham: Springer 373-83. https://doi.org/10.1007/978-3-319-44174-0_23
|
| [88] |
Ali R, Goubran M, Choudhri O, et al. (2015) Seven-Tesla MRI and neuroimaging biomarkers for Alzheimer's disease. Neurosurg Focus 39: E4. https://doi.org/10.3171/2015.9.FOCUS15326
|
| [89] |
van Gemert J, Brink W, Webb A, et al. (2019) High-permittivity pad design tool for 7T neuroimaging and 3T body imaging. Magn Reson Med 81: 3370-3378. https://doi.org/10.1002/mrm.27629
|
| [90] |
Uwano I, Kudo K, Yamashita F, et al. (2014) Intensity inhomogeneity correction for magnetic resonance imaging of human brain at 7T. Med Phys 41: 022302. https://doi.org/10.1118/1.4860954
|
| [91] |
Truong TK, Chakeres DW, Beversdorf DQ, et al. (2006) Effects of static and radiofrequency magnetic field inhomogeneity in ultra-high field magnetic resonance imaging. Magn Reson Imaging 24: 103-112. https://doi.org/10.1016/j.mri.2005.09.013
|
| [92] |
Trattnig S, Springer E, Bogner W, et al. (2018) Key clinical benefits of neuroimaging at 7T. NeuroImage 168: 477-489. https://doi.org/10.1016/j.neuroimage.2016.11.031
|
| [93] |
van Gelderen P, de Zwart JA, Starewicz P, et al. (2007) Real-time shimming to compensate for respiration-induced B0 fluctuations. Magn Reson Med 57: 362-368. https://doi.org/10.1002/mrm.211369
|
| [94] |
Versluis MJ, Peeters JM, van Rooden S, et al. (2010) Origin and reduction of motion and f0 artifacts in high resolution T2*-weighted magnetic resonance imaging: application in Alzheimer's disease patients. NeuroImage 51: 1082-1088. https://doi.org/10.1016/j.neuroimage.2010.03.048
|
| [95] | Perera Molligoda Arachchige AS (2023) Neuroimaging with PET/MR: moving beyond 3T in preclinical systems, when for clinical practice?. Clin Trans Imaging (Springer) . Advance online publication |
| [96] |
Yuan Y, Gu ZX, Wei WS (2009) Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: a meta-analysis. Am J Neuroradiol 30: 404-410. https://doi.org/10.3174/ajnr.A1357
|
| [97] |
Perera Molligoda Arachchige AS (2021) What must be done in case of a dense collection?. Radiol Med 126: 1657-1658. https://doi.org/10.1007/s11547-021-01426-9
|
| [98] | Verma Y, Ramesh S, Perera Molligoda Arachchige AS (2023) 7 T Versus 3 T in the Diagnosis of Small Unruptured Intracranial Aneurysms: Reply to Radojewski et al. Clin Neuroradiol . https://doi.org/10.1007/s00062-023-01321-y |

Figures(6)

Arosh S. Perera Molligoda Arachchige, Anton Kristoffer Garner. Seven Tesla MRI in Alzheimer's disease research: State of the art and future directions: A narrative review[J]. AIMS Neuroscience, 2023, 10(4): 401-422. doi: 10.3934/Neuroscience.2023030







 DownLoad:
DownLoad: