Citation: Issam Tout, Marie Marotel, Isabelle Chemin, Uzma Hasan. HBV and the importance of TLR9 on B cell responses[J]. AIMS Allergy and Immunology, 2017, 1(3): 124-137. doi: 10.3934/Allergy.2017.3.124

| [1] | Hepatitis B (2017) Available from: http://www.who.int/mediacentre/factsheets/fs204_Jul2014/en/. |
| [2] | Lavanchy D (2005) Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J Clin Virol 34: S1–S3. |
| [3] |
McMahon BJ (2005) Epidemiology and natural history of hepatitis B. Semin Liver Dis 25: 3–8. doi: 10.1055/s-2005-915644

|
| [4] | Bertoletti A, Gehring AJ (2006) The immune response during hepatitis B virus infection. J Gen Viro l87: 1439–1449. |
| [5] |
Dienstag JL (2008) Hepatitis B virus infection. New Engl J Med 359: 1486–1500. doi: 10.1056/NEJMra0801644
|
| [6] | Mason WS, Aldrich C, Summers J, et al. (1982) Asymmetric replication of duck hepatitis B virus DNA in liver cells: free minus-strand DNA. Proc Natl Acad Sci USA 79: 3997–4001. |
| [7] |
Summers J, Mason WS (1982) Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29: 403–415. doi: 10.1016/0092-8674(82)90157-X
|
| [8] | Ganem D (2001) Virology. The X files--one step closer to closure. Science 294: 2299–2300. |
| [9] |
European Association For The Study Of The Liver (2012) EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol 57: 167–185. doi: 10.1016/j.jhep.2012.02.010
|
| [10] |
Bertoletti A, Kennedy PT (2015) The immune tolerant phase of chronic HBV infection: new perspectives on an old concept. Cell Mol Immunol 12: 258–263. doi: 10.1038/cmi.2014.79
|
| [11] |
Bertoletti A, Ferrari C (2013) Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Postgrad Med J 89: 294–304. doi: 10.1136/postgradmedj-2011-301073rep
|
| [12] |
Guidotti LG, Chisari FV (2006) Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol 1: 23–61. doi: 10.1146/annurev.pathol.1.110304.100230
|
| [13] |
McMahon BJ (2009) The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol Int 3: 334–342. doi: 10.1007/s12072-008-9112-z
|
| [14] | Buti M, Rodriguez-Frias F, Jardi R, et al. (2005) Hepatitis B virus genome variability and disease progression: the impact of pre-core mutants and HBV genotypes. J Clin Virol 34: S79–S82. |
| [15] |
Sonneveld MJ, Rijckborst V, Zeuzem S, et al. (2012) Presence of precore and core promoter mutants limits the probability of response to peginterferon in hepatitis B e antigen-positive chronic hepatitis B. Hepatology 56: 67–75. doi: 10.1002/hep.25636
|
| [16] | Tacke F, Gehrke C, Luedde T, et al. (2004) Basal core promoter and precore mutations in the hepatitis B virus genome enhance replication efficacy of Lamivudine-resistant mutants. J Virol 78: 8524–8535. |
| [17] |
Alberti A, Diana S, Sculard GH, et al. (1978) Detection of a new antibody system reacting with Dane particles in hepatitis B virus infection. BMJ-Brit Med J 2: 1056–1058. doi: 10.1136/bmj.2.6144.1056
|
| [18] | Loggi E, Gamal N, Bihl F, et al. (2014) Adaptive response in hepatitis B virus infection. J Viral Hepatitis 21: 305–313. |
| [19] |
Hangartner L, Zinkernagel RM, Hengartner H (2006) Antiviral antibody responses: the two extremes of a wide spectrum. Nat Rev Immunol 6: 231–243. doi: 10.1038/nri1783
|
| [20] |
Seeger C, Mason WS (2000) Hepatitis B virus biology. Microbiol Mol Biol R 64: 51–68. doi: 10.1128/MMBR.64.1.51-68.2000
|
| [21] | Weber B (2005) Genetic variability of the S gene of hepatitis B virus: clinical and diagnostic impact. J Clin Virol 32: 102–112. |
| [22] | Sayan M, Sentürk O, Akhan SÇ, et al. (2010) Monitoring of hepatitis B virus surface antigen escape mutations and concomitantly nucleos(t)ide analog resistance mutations in Turkish patients with chronic hepatitis B. Int J Infect Dis 14: e136–e141. |
| [23] |
Mitchison NA (2004) T-cell-B-cell cooperation. Nat Rev Immunol 4: 308–312. doi: 10.1038/nri1334
|
| [24] |
Garraud O, Borhis G, Badr G, et al. (2012) Revisiting the B-cell compartment in mouse and humans: more than one B-cell subset exists in the marginal zone and beyond. BMC Immunol 13: 63. doi: 10.1186/1471-2172-13-63
|
| [25] |
LeBien TW, Tedder TF (2008) B lymphocytes: how they develop and function. Blood 112: 1570–1580. doi: 10.1182/blood-2008-02-078071
|
| [26] | Das A, Ellis G, Pallant C, et al. (2012) IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol 189: 3925–3935. |
| [27] |
Xu X, Shang Q, Chen X, et al. (2015) Reversal of B-cell hyperactivation and functional impairment is associated with HBsAg seroconversion in chronic hepatitis B patients. Cell Mol Immunol 12: 309–316. doi: 10.1038/cmi.2015.25
|
| [28] |
Liu Y, Cheng LS, Wu S, et al. (2016) IL-10-producing regulatory B-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin Sci 130: 907–919. doi: 10.1042/CS20160069
|
| [29] |
Farci P, Diaz G, Chen Z, et al. (2010) B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA 107: 8766–8771. doi: 10.1073/pnas.1003854107
|
| [30] |
Moir S, Ho J, Malaspina A, et al. (2008) Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med 205: 1797–1805. doi: 10.1084/jem.20072683
|
| [31] | Weiss GE, Crompton PD, Li S, et al. (2009) Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol 183: 2176–2182. |
| [32] |
Oliviero B, Cerino A, Varchetta S, et al. (2011) Enhanced B-cell differentiation and reduced proliferative capacity in chronic hepatitis C and chronic hepatitis B virus infections. J Hepatol 55: 53–60. doi: 10.1016/j.jhep.2010.10.016
|
| [33] |
Moir S, Fauci AS (2009) B cells in HIV infection and disease. Nat Rev Immunol 9: 235–245. doi: 10.1038/nri2524
|
| [34] |
Suarez F, Lortholary O, Hermine O, et al. (2006) Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood 107: 3034–3044. doi: 10.1182/blood-2005-09-3679
|
| [35] |
Maruyama T, McLachlan A, Iino S, et al. (1993) The serology of chronic hepatitis B infection revisited. J Clin Invest 91: 2586–2595. doi: 10.1172/JCI116497
|
| [36] |
Böcher WO, Herzog-Hauff S, Herr W, et al. (1996) Regulation of the neutralizing anti-hepatitis B surface (HBs) antibody response in vitro in HBs vaccine recipients and patients with acute or chronic hepatitis B virus (HBV) infection. Clin Exp Immunol 105: 52–58. doi: 10.1046/j.1365-2249.1996.d01-732.x
|
| [37] |
Dusheiko GM, Hoofnagle JH, Cooksley WG, et al. (1983) Synthesis of antibodies to hepatitis B virus by cultured lymphocytes from chronic hepatitis B surface antigen carriers. J Clin Invest 71: 1104–1113. doi: 10.1172/JCI110860
|
| [38] | Barnaba V, Valesini G, Levrero M, et al. (1985) Immunoregulation of the in vitro anti-HBs antibody synthesis in chronic HBsAg carriers and in recently boosted anti-hepatitis B vaccine recipients. Clin Exp Immunol 60: 259–266. |
| [39] | Barnaba V, Levrero M, Ruberti G, et al. (1987) In vitro anti-HBs antibody synthesis from anti-hepatitis B vaccine recipients. Clin Exp Immunol 70: 283–288. |
| [40] | Amu S, Ruffin N, Rethi B, et al. (2013) Impairment of B-cell functions during HIV-1 infection. Aids 27: 2323–2334. |
| [41] | Pensieroso S, Galli L, Nozza S, et al. (2013) B-cell subset alterations and correlated factors in HIV-1 infection. Aids 27: 1209–1217. |
| [42] | Ni J, Hembrador E, Di BA, et al. (2003) Accumulation of B lymphocytes with a naive, resting phenotype in a subset of hepatitis C patients. J Immunol 170: 3429–3439. |
| [43] |
Racanelli V, Frassanito MA, Leone P, et al. (2006) Antibody production and in vitro behavior of CD27-defined B-cell subsets: persistent hepatitis C virus infection changes the rules. J Virol 80: 3923–3934. doi: 10.1128/JVI.80.8.3923-3934.2006
|
| [44] |
Rosa D, Saletti G, Gregorio ED, et al. (2005) Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci USA 102: 18544–18549. doi: 10.1073/pnas.0509402102
|
| [45] |
Wang XD, Wang L, Ji FJ, et al. (2012) Decreased CD27 on B lymphocytes in patients with primary hepatocellular carcinoma. J Int Med Res 40: 307–316. doi: 10.1177/147323001204000131
|
| [46] | Li WY, Jiang YF, Jin QL, et al. (2010) Immunologic characterization of posthepatitis cirrhosis caused by HBV and HCV infection. J Biomed Biotechnol 2010: 138237. |
| [47] |
Lund FE (2008) Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 20: 332–338. doi: 10.1016/j.coi.2008.03.003
|
| [48] |
Llorente L, Zou W, Levy Y, et al. (1995) Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J Exp Med 181: 839–844. doi: 10.1084/jem.181.3.839
|
| [49] | Kakumu S, Shinagawa T, Ishikawa T, et al. (1991) Serum interleukin 6 levels in patients with chronic hepatitis B. Am J Gastroenterol 86: 1804–1808. |
| [50] |
Aspord C, Bruder CJ, Jacob MC, et al. (2016) Remodeling of B-cell subsets in blood during pegylated IFNα-2a therapy in patients with chronic Hepatitis B infection. PloS One 11: e0156200. doi: 10.1371/journal.pone.0156200
|
| [51] |
Chang JJ, Wightman F, Bartholomeusz A, et al. (2005) Reduced hepatitis B virus (HBV)-specific CD4+ T-cell responses in human immunodeficiency virus type 1-HBV-coinfected individuals receiving HBV-active antiretroviral therapy. J Virol 79: 3038–3051. doi: 10.1128/JVI.79.5.3038-3051.2005
|
| [52] |
Maini MK, Boni C, Lee CK, et al. (2000) The role of virus-specific CD8 (+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med 191: 1269–1280. doi: 10.1084/jem.191.8.1269
|
| [53] | Boni C, Penna A, Ogg GS, et al. (2001) Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology 33: 963–971. |
| [54] | Chen L, Zhang Z, Chen W, et al. (2007) B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J Immunol 178: 6634–6641. |
| [55] |
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820. doi: 10.1016/j.cell.2010.01.022
|
| [56] |
Hua Z, Hou B (2013) TLR signaling in B-cell development and activation. Cell Mol Immunol 10: 103–106. doi: 10.1038/cmi.2012.61
|
| [57] |
Bernasconi NL, Onai N, Lanzavecchia A (2003) A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 101: 4500–4504. doi: 10.1182/blood-2002-11-3569
|
| [58] | Ruprecht CR, Lanzavecchia A (2006) Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol 36: 810–816. |
| [59] |
Hirsch I, Caux C, Hasan U, et al. (2010) Impaired Toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol 31: 391–397. doi: 10.1016/j.it.2010.07.004
|
| [60] | Chen Z, Cheng Y, Xu Y, et al. (2008) Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol 128: 400–408. |
| [61] | Fathallah I, Parroche P, Gruffat H, et al. (2010) EBV latent membrane protein 1 is a negative regulator of TLR9. J Immunol 185: 6439–6447. |
| [62] |
Hasan UA, Zannetti C, Parroche P, et al. (2013) The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J Exp Med 210: 1369–1387. doi: 10.1084/jem.20122394
|
| [63] |
Wieland S, Thimme R, Purcell RH, et al. (2004) Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA 101: 6669–6674. doi: 10.1073/pnas.0401771101
|
| [64] |
Dunn C, Peppa D, Khanna P, et al. (2009) Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 137: 1289–1300. doi: 10.1053/j.gastro.2009.06.054
|
| [65] |
Cheng J, Imanishi H, Morisaki H, et al. (2005) Recombinant HBsAg inhibits LPS-induced COX-2 expression and IL-18 production by interfering with the NFkappaB pathway in a human monocytic cell line, THP-1. J Hepatol 43: 465–471. doi: 10.1016/j.jhep.2005.02.033
|
| [66] | Müller C, Zielinski CC (1990) Impaired lipopolysaccharide-inducible tumor necrosis factor production in vitro by peripheral blood monocytes of patients with viral hepatitis. Hepatology 12: 1118–1124. |
| [67] | Wang S, Chen Z, Hu C, et al. (2013) Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol 190: 5142–5151. |
| [68] |
Ml ODB, Binda RS, van Roosmalen MH, et al. (2009) Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology 126: 280–289. doi: 10.1111/j.1365-2567.2008.02896.x
|
| [69] |
Vincent IE, Zannetti C, Lucifora J, et al. (2011) Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PloS One 6: e26315. doi: 10.1371/journal.pone.0026315
|
| [70] |
Xu Y, Hu Y, Shi B, et al. (2009) HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol Immunol 46: 2640–2646. doi: 10.1016/j.molimm.2009.04.031
|
| [71] |
Han Q, Zhang C, Zhang J, et al. (2013) The role of innate immunity in HBV infection. Semin Immunopathol 35: 23–38. doi: 10.1007/s00281-012-0331-y
|
| [72] |
Lang T, Lo C, Skinner N, et al. (2011) The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol 55: 762–769. doi: 10.1016/j.jhep.2010.12.042
|
| [73] |
Visvanathan K, Skinner NA, Thompson AJV, et al. (2007) Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 45: 102–110. doi: 10.1002/hep.21482
|
| [74] | Wu J, Meng Z, Jiang M, et al. (2009) Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 49: 1132–1140. |
| [75] |
Luangsay S, Gruffaz M, Isorce N, et al. (2015) Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol 63: 1314–1322. doi: 10.1016/j.jhep.2015.07.014
|
| [76] |
Jegaskanda S, Ahn SH, Skinner N, et al. (2014) Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J Virol 88: 10412–10420. doi: 10.1128/JVI.00111-14
|
| [77] |
Yu S, Chen J, Wu M, et al. (2010) Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol 91: 2080–2090. doi: 10.1099/vir.0.020552-0
|
| [78] | Hepatitis B virus polymerase suppresses NF-κB signaling by inhibiting the activity of IKKs via interaction with Hsp90β. Available from: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pubmed?term=Hepatitis%20B%20virus%20polymerase%20suppresses%20NF-kappaB%20signaling%20by%20inhibiting%20the%20activity%20of%20IKKs%20via%20interaction%20with%20Hsp90beta%5BTitle%5D. |
| [79] |
Wang H, Ryu WS (2010) Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 6: e1000986. doi: 10.1371/journal.ppat.1000986
|
| [80] |
Martinet J, Dufeu-Duchesne T, Bruder CJ, et al. (2012) Altered functions of plasmacytoid dendritic cells and reduced cytolytic activity of natural killer cells in patients with chronic HBV infection. Gastroenterology 143: 1586–1596. doi: 10.1053/j.gastro.2012.08.046
|
| [81] | Xu N, Yao HP, Lv GC, et al. (2012) Downregulation of TLR7/9 leads to deficient production of IFN-α from plasmacytoid dendritic cells in chronic hepatitis B. Inflamm Res 61: 997–1004. |
| [82] |
Ratnam DT, Sievert W, Visvanathan K (2012) Natural killer cells display impaired responses to toll like receptor 9 that support viral persistence in chronic hepatitis B. Cell Immunol 279: 109–115. doi: 10.1016/j.cellimm.2012.09.005
|
| [83] |
Wang K, Liu H, He Y, et al. (2010) Correlation of TLR1-10 expression in peripheral blood mononuclear cells with chronic hepatitis B and chronic hepatitis B-related liver failure. Hum Immunol 71: 950–956. doi: 10.1016/j.humimm.2010.07.013
|
| [84] |
Zhou J, Huang Y, Tian D, et al. (2009) Expression of toll-like receptor 9 in peripheral blood mononuclear cells from patients with different hepatitis B and C viral loads. J Huazhong University Sci Technol Med Sci 29: 313–317. doi: 10.1007/s11596-009-0310-2
|
| [85] | Xie Q, Shen HC, Jia NN, et al. (2009) Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect 11: 515–523. |
| [86] |
Huang YW, Hsu CK, Lin SC, et al. (2014) Reduced toll-like receptor 9 expression on peripheral CD14+ monocytes of chronic hepatitis B patients and its restoration by effective therapy. Antivir Ther 19: 637–643. doi: 10.3851/IMP2762
|
| [87] |
Xu N, Yao H, Sun Z, et al. (2008) Toll-like receptor 7 and 9 expression in peripheral blood mononuclear cells from patients with chronic hepatitis B and related hepatocellular carcinoma. Acta Pharmacol Sin 29: 239–244. doi: 10.1111/j.1745-7254.2008.00711.x
|
| [88] | Zhao PW, Ma L, Ji HF, et al. (2015) The expression of TLR-9, CD86, and CD95 phenotypes in circulating B cells of patients with chronic viral hepatitis B or C before and after antiviral therapy. Mediat Inflamm 2015: 762709. |
| [89] | Wang G, Liu Y, Huang R, et al. (2017) Characteristics of regulatory B cells in patients with chronic hepatitis B virus infection in different immune phases. Discov Med 23: 295–304. |
| [90] | Takeshita F, Suzuki K, Sasaki S, et al. (2004) Transcriptional regulation of the human TLR9 gene. J Immunol 173: 2552–2561. |
| [91] | Aoyama T, Paik YH, Seki E (2010) Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract 2010: E1–E8. |
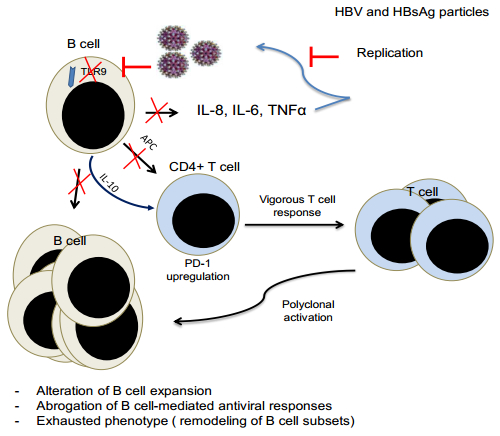
Figures(1) / Tables(1)

Issam Tout, Marie Marotel, Isabelle Chemin, Uzma Hasan. HBV and the importance of TLR9 on B cell responses[J]. AIMS Allergy and Immunology, 2017, 1(3): 124-137. doi: 10.3934/Allergy.2017.3.124







 DownLoad:
DownLoad: