Citation: Yves Achdou, Ziad Kobeissi. Mean field games of controls: Finite difference approximations[J]. Mathematics in Engineering, 2021, 3(3): 1-35. doi: 10.3934/mine.2021024

| [1] | UMFPACK. Available from: http://www.cise.ufl.edu/research/sparse/umfpack/current/. |
| [2] | Achdou Y (2013) Finite difference methods for mean field games, In: Hamilton-Jacobi Equations: Approximations, Numerical Analysis and Applications, Heidelberg: Springer, 1-47. |
| [3] | Achdou Y, Capuzzo-Dolcetta I (2010) Mean field games: Numerical methods. SIAM J Numer Anal 48: 1136-1162. |
| [4] | Achdou Y, Lasry JM (2019) Mean field games for modeling crowd motion, In: Contributions to Partial Differential Equations and Applications, Cham: Springer, 17-42. |
| [5] | Achdou Y, Porretta A (2018) Mean field games with congestion. Ann I H Poincaré Anal non linéaire 35: 443-480, |
| [6] | Bensoussan A, Frehse J, Yam P (2013) Mean field games and mean field type control theory. New York: Springer. |
| [7] | Bonnans FJ, Hadikhanloo S, Pfeiffer L (2019) Schauder estimates for a class of potential mean field games of controls. arXiv: 1902.05461. |
| [8] | Cardaliaguet P, Lehalle CA (2018) Mean field game of controls and an application to trade crowding. Math Financ Econ 12: 335-363. |
| [9] | Carmona R, Delarue F (2015) Forward-backward stochastic differential equations and controlled McKean-Vlasov dynamics. Ann Probab 43: 2647-2700. |
| [10] | Carmona R, Delarue F (2018) Probabilistic theory of mean field games with applications I, Cham: Springer. |
| [11] | Carmona R, Lacker D (2015)) A probabilistic weak formulation of mean field games and applications. Ann Appl Probab 25: 1189-1231. |
| [12] | Gomes D, Voskanyan V (2013) Extended mean field games. Izv Nats Akad Nauk Armenii Mat 48: 63-76. |
| [13] | Gomes DA, Patrizi S, Voskanyan V (2014) On the existence of classical solutions for stationary extended mean field games. Nonlinear Anal 99: 49-79. |
| [14] | Huang M, Caines PE, Malhamé RP (2007) Large-population cost-coupled LQG problems with nonuniform agents: Individual-mass behavior and decentralized $\epsilon$-Nash equilibria. IEEE T Automat Contr 52: 1560-1571. |
| [15] | Huang M, Caines PE, Malhamé RP (2006) Large population stochastic dynamic games: closed-loop McKean-Vlasov systems and the Nash certainty equivalence principle. Commun Inf Syst 6: 221-251. |
| [16] | Kobeissi Z (2019) On Classical Solutions to the Mean Field Game System of Controls. arXiv: 1904.11292. |
| [17] | Lasry JM, Lions PL (2006) Jeux à champ moyen. I. Le cas stationnaire. C R Math Acad Sci Paris 343: 619-625. |
| [18] | Lasry JM, Lions PL (2006) Jeux à champ moyen. II. Horizon fini et contr?le optimal. C R Math Acad Sci Paris 343: 679-684. |
| [19] | Lasry JM, Lions PL (2007) Mean field games. JPN J Math 2: 229-260. |
| [20] | Lions PL, Théorie des jeux à champs moyen. Video lecture series at Collège de France, 2011-2019. Available from: https://www.college-de-france.fr/site/pierre-louis-lions/index.htm. |
| [21] | van der Vorst HA (1992) Bi-CGSTAB: A fast and smoothly converging variant of Bi-CG for the solution of nonsymmetric linear systems. SIAM J Sci Statist Comput 13: 631-644. |
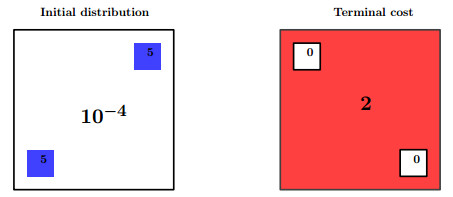
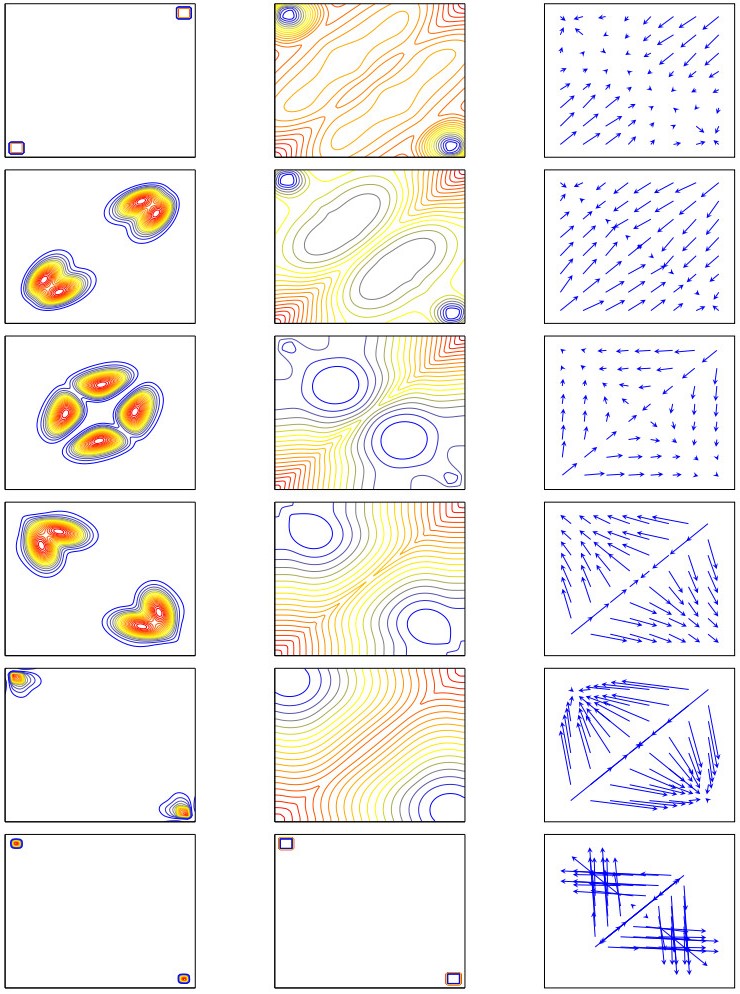
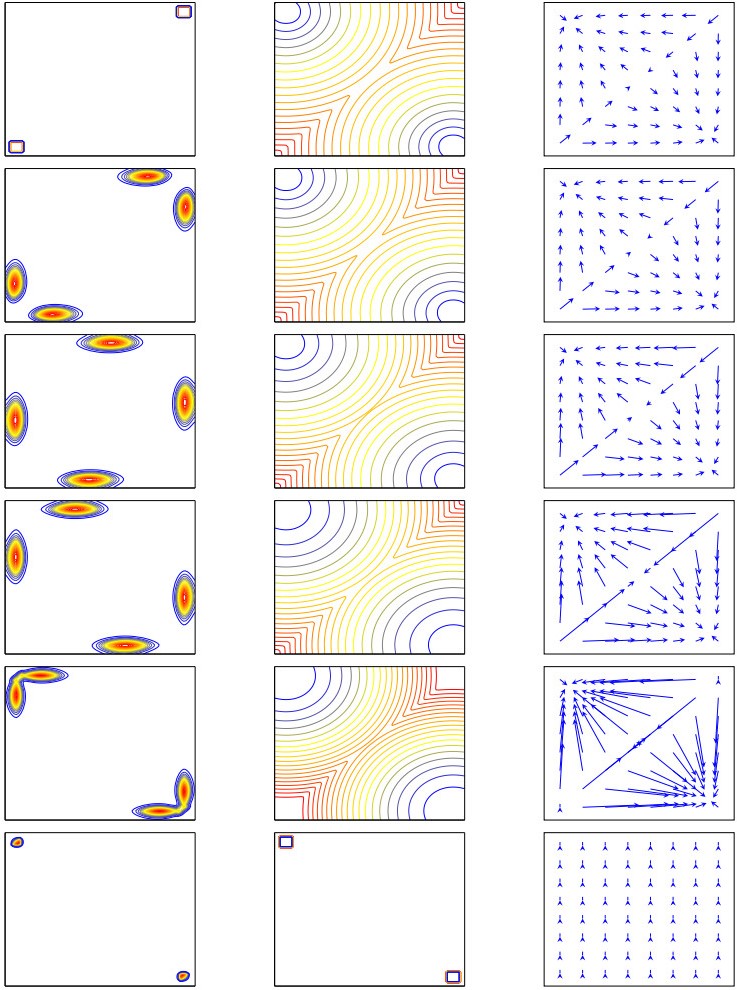
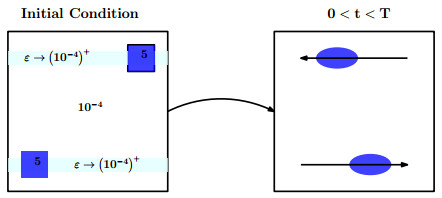
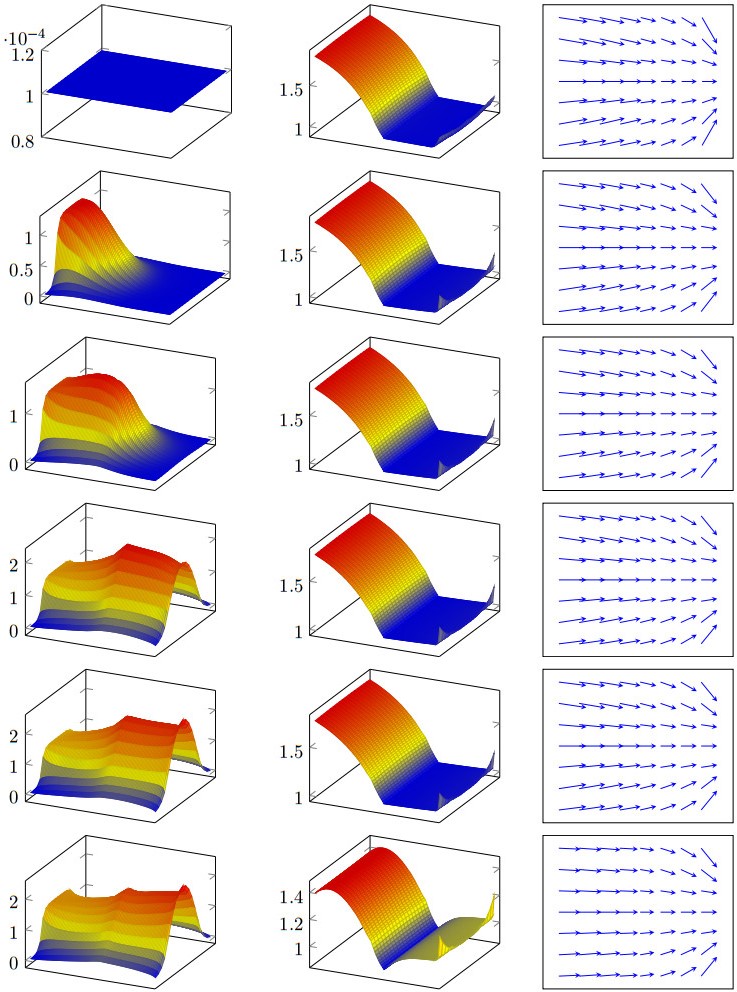
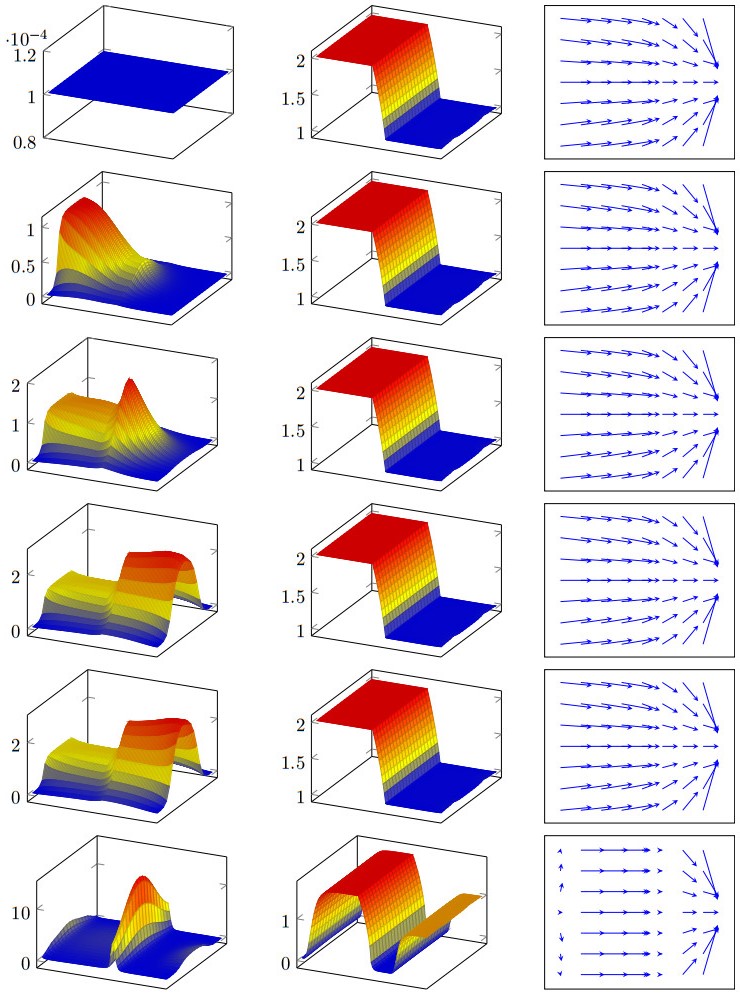
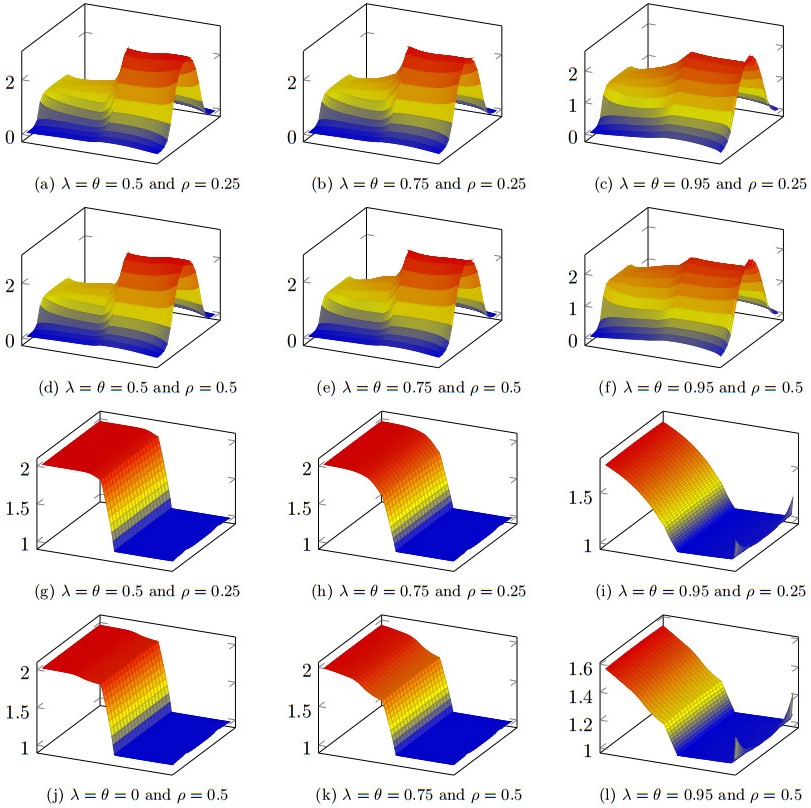
Figures(11) / Tables(3)

Yves Achdou, Ziad Kobeissi. Mean field games of controls: Finite difference approximations[J]. Mathematics in Engineering, 2021, 3(3): 1-35. doi: 10.3934/mine.2021024







 DownLoad:
DownLoad: