Citation: Carlos Gutierrez-Merino, Dorinda Marques-da-Silva, Sofia Fortalezas, Alejandro K. Samhan-Arias. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracellular potassium deprivation[J]. AIMS Molecular Science, 2016, 3(1): 12-29. doi: 10.3934/molsci.2016.1.12

| [1] |
Contestabile A (2002) Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. The Cerebellum 1: 41-55. doi: 10.1080/147342202753203087

|
| [2] | Gallo V, Kingsbury A, Balazs R, et al. (1987) The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci 7: 2203-2213. |
| [3] |
Balazs R, Gallo V, Kingsbury A (1988) Effect of depolarization on the maturation of cerebellar granule cells in culture. Devel Brain Res 40: 269-276. doi: 10.1016/0165-3806(88)90139-3
|
| [4] |
D’Mello SR, Galli C, Ciotti T, et al. (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989-10993. doi: 10.1073/pnas.90.23.10989
|
| [5] | Copani A, Bruno VMG, Barresi V, et al. (1994) Activation of metabotropic glutamate receptors prevents neuronal apoptosis in culture. J Neurochem 64: 101-108. |
| [6] |
Ciani E, Rizzi S, Paulsen RE, et al. (1997) Chronic pre-explant blockade of the NMDA receptor affects survival of cerebellar granule cells explanted in vitro. Devel Brain Res 99: 112-117. doi: 10.1016/S0165-3806(96)00187-3
|
| [7] | Sparapani M, Virgili M, Bardi G (1998) Ornithine decarboxylase activity during development of cerebellar granule neurons. J Neurochem 71: 1898-1904. |
| [8] | Martin-Romero FJ, Garcia-Martin E, Gutierrez-Merino C (1996) Involvement of free radicals in signaling of low-potassium induced apoptosis in cultured cerebellar granule cells. Int J Dev Biol Suppl.1: 197S-198S. |
| [9] |
Martin-Romero FJ, Garcia-Martin E, Gutierrez-Merino C (2002) Inhibition of the oxidative stress produced by plasma membrane NADH oxidase delays low-potassium induced apoptosis of cerebellar granule cells. J Neurochem 82: 705-715. doi: 10.1046/j.1471-4159.2002.01023.x
|
| [10] | Nardi N, Avidan G, Daily D, et al. (1997) Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem 68: 750-759. |
| [11] | Schulz JB, Beinroth S, Weller M, et al. (1998) Endonucleolytic DNA fragmentation is not required for apoptosis of cultured rat cerebellar granule neurons. Neurosci Lett 27: 9-12. |
| [12] | Marks N, Berg MJ, Guidotti A, et al. (1998) Activation of caspase-3 and apoptosis in cerebellar granule cells. J Neurosci Res 52: 334-341. |
| [13] |
Allsopp TE, McLuckie J, Kerr LE, et al. (2000) Caspase 6 activity initiates caspase 3 activation in cerebellar granule cell apoptosis. Cell Death Differ 7: 984-993. doi: 10.1038/sj.cdd.4400733
|
| [14] | Eldadah BA, Ren RF, Faden AI (2000) Ribozyme-mediated inhibition of caspase-3 protects cerebellar granule cells from apoptosis induced by serum-potassium deprivation. J Neurosci 20: 179-186. |
| [15] |
Cowling V, Downward J (2002) Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ 9: 1046-1056. doi: 10.1038/sj.cdd.4401065
|
| [16] |
Valencia A, Morán J (2001) Role of oxidative stress in the apoptotic cell death of cultured cerebellar granule neurons. J Neurosci Res 64: 284-297. doi: 10.1002/jnr.1077
|
| [17] | Simons M, Beinroth S, Gleichmann M, et al. (1999) Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J Neurochem 72: 292-301. |
| [18] | Wigdal SS, Kirkland RA, Franklin JL, et al. (2002) Cytochrome c release precedes mitochondrial membrane potential loss in cerebellar granule neurons apoptosis: lack of mitochondrial swelling. J Neurochem 82: 1029-1038. |
| [19] |
Samhan-Arias AK, Marques-da-Silva D, Yanamala N, et al. (2012) Stimulation and clustering of cytochrome b5 reductase in caveolin-rich lipid microdomains is an early event in oxidative stress-mediated apoptosis of cerebellar granule neurons. J Proteomics 75: 2934-2949. doi: 10.1016/j.jprot.2011.12.007
|
| [20] |
Bobba A, Atlante A, Giannattasio S, et al. (1999) Early release and subsequent caspase-mediated degradation of cytochrome c in apoptotic cerebellar granule neurons. FEBS Lett 457: 126-130. doi: 10.1016/S0014-5793(99)01018-2
|
| [21] | McGinnis KM, Gnegy ME, Wang KK (1999) Endogenous bax translocation in SH-SY5Y human neuroblastoma cells and cerebellar granule neurons undergoing apoptosis. J Neurochem 72: 1899-1906. |
| [22] |
Miller TM, Moulder KL, Knudson CM, et al. (1997) Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol 139: 205-217. doi: 10.1083/jcb.139.1.205
|
| [23] | Cregan SP, MacLaurin JG, Craig CG, et al. (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci 19: 7860-7869. |
| [24] |
Tanabe H, Eguchi Y, Kamada S, et al. (1997) Susceptibility of cerebellar granule neurons derived from Bcl-2- deficient and transgenic mice to cell death. Eur J Neurosci 9: 848-856. doi: 10.1111/j.1460-9568.1997.tb01434.x
|
| [25] |
Gleichmann M, Beinroth S, Reed JC, et al. (1998) Potassium deprivation-induced apoptosis of cerebellar granule neurons: cytochrome c release in the absence of altered expression of Bcl-2 family proteins. Cell Physiol Biochem 8: 194-201. doi: 10.1159/000016282
|
| [26] | Galli C, Meucci O, Scorziello A, et al. (1995) Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin and IGF-I through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci 15: 1172-1179. |
| [27] |
Kubo T, Nonomura T, Enokido Y, et al. (1995) Brain derived neurotrophic factor (BDNF) can prevent apoptosis of rat cerebellar granule neurons in culture. Devel Brain Res 85: 249-258. doi: 10.1016/0165-3806(94)00220-T
|
| [28] |
Chang JY, Korolev VV, Wang JZ (1996) Cyclic AMP and pituitary adenylate cyclase-activating polypeptide (PACAP) prevent programmed cell death of cultured cerebellar granule cells. Neurosci Lett 206: 181-184. doi: 10.1016/S0304-3940(96)12468-X
|
| [29] |
Campard PK, Crochemore C, Rene F, et al. (1997) PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol 16: 323-333. doi: 10.1089/dna.1997.16.323
|
| [30] | Ikeuchi T, Shimoke K, Kubo T, et al. (1998) Apoptosis- inducing and -preventing signal transduction pathways in cultured cerebellar granule neurons. Hum Cell 11: 125-140. |
| [31] |
Koulich E, Nguyen T, Johnson K, et al. (2001) NFkappaB is involved in the survival of cerebellar granule neurons: association of NF-kappabeta phosphorylation with cell survival. J Neurochem 76: 1188-1198. doi: 10.1046/j.1471-4159.2001.00134.x
|
| [32] | D’Mello SR, Borodezt K, Soltoff SP (1997) Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-I signaling. J Neurosci 17: 1548-1560. |
| [33] |
Dudek H, Datta SR, Franke TF, et al. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665. doi: 10.1126/science.275.5300.661
|
| [34] |
Shimoke K, Kubo T, Numakawa T, et al. (1997) Involvement of phosphatidylinositol- 3 kinase in prevention of low K+-induced apoptosis of cerebellar granule neurons. Devel Brain Res 101: 197-206. doi: 10.1016/S0165-3806(97)00065-5
|
| [35] | Bhave SV, Ghoda L, Hoffman PL (1999) Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascade and site of ethanol action. J Neurosci 19: 3277-3286. |
| [36] |
Shimoke K, Yamagishi S, Yamada M, et al. (1999) Inhibition of phosphatidylinositol 3-kinase activity elevates c-Jun N-terminal kinase activity in apoptosis of cultured cerebellar granule neurons. Devel Brain Res 112: 245-253. doi: 10.1016/S0165-3806(98)00172-2
|
| [37] |
Le-Niculescu H, Bonfoco E, Kasuya Y, et al. (1999) Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol 19: 751-763. doi: 10.1128/MCB.19.1.751
|
| [38] | Vaudry D, Gonzalez BJ, Basille M, et al. (2000) PACAP acts as a neurotrophic factor during histogenesis of the rat cerebellar cortex. Ann N Y Acad Sci 921: 293-299. |
| [39] | Cavallaro S, Copani A, D’Agata V, et al. (1996) Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol Pharmacol 50: 60-66. |
| [40] | Villalba M, Bockaert J, Journot L (1997) Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci 17: 83-90. |
| [41] |
Journot L, Villalba M, Bockaert J (1998) PACAP-38 protects cerebellar granule cells from apoptosis. Ann N Y Acad Sci 865: 100-110. doi: 10.1111/j.1749-6632.1998.tb11168.x
|
| [42] |
Franklin JL, Johnson Jr EM (1992) Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci 15: 501-508. doi: 10.1016/0166-2236(92)90103-F
|
| [43] |
Franklin JL, Johnson Jr EM (1994) Block of neuronal apoptosis by a sustained increase of steady-state free Ca2+ concentration. Philos Trans R Soc Lond B Biol Sci 345: 251-256. doi: 10.1098/rstb.1994.0102
|
| [44] |
Gutierrez-Martin Y, Martin-Romero FJ, Henao F, et al. (2005) Alteration of cytosolic free calcium homeostasis by SIN-1: high sensitivity of L-type Ca2+ channels to extracellular oxidative/nitrosative stress in cerebellar granule cells. J Neurochem 92: 973-989. doi: 10.1111/j.1471-4159.2004.02964.x
|
| [45] |
Copani A, Casabona V, Bruno A, et al. (1998) The metabotropic glutamate receptor mGlu5 controls the onset of developmental apoptosis in cultured cerebellar neurons. Eur J Neurosci 10: 2173-2184. doi: 10.1046/j.1460-9568.1998.00230.x
|
| [46] | Borodetz K, D’Mello SRD (1998) Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J Neurosci Res 53: 531-541. |
| [47] |
Garcia-Bereguiain MA, Samhan-Arias AK, Martin-Romero FJ, et al. (2008) Hydrogen sulfide raises cytosolic calcium in neurons through activation of L-type Ca2+ channels. Antioxid Redox Signal 10: 31-42. doi: 10.1089/ars.2007.1656
|
| [48] |
Balazs R, Jorgensen OS, Hack N (1988) N-methyl-D-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience 27: 437-451. doi: 10.1016/0306-4522(88)90279-5
|
| [49] |
Balazs A, Hack N, Jorgensen OS (1990) Selective stimulation of excitatory amino acid receptor subtypes and the survival of cerebellar granule cells in culture: Effect of kainic acid. Neuroscience 37: 251-258. doi: 10.1016/0306-4522(90)90211-L
|
| [50] |
Balazs R, Hack N, Jorgensen OS (1990) Interactive effects involving different classes of excitatory amino acid receptors and the survival of cerebellar granule cells in culture. Int J Devel Neurosci 8: 347-359. doi: 10.1016/0736-5748(90)90068-D
|
| [51] | Yan GM, Lin SZ, Irwin RP, et al. (1995) Activation of muscarinic cholinergic receptor blocks apoptosis of cultured cerebellar granule neurons. Mol Pharmacol 47: 248-257. |
| [52] | Mattson MP (1996) Calcium and free radicals: mediators of neurotrophic factor and excitatory transmitter-regulated developmental plasticity and cell death. Perspect Dev Neurobiol 3: 79-91. |
| [53] |
Altman J (1982) Morphological development of the rat cerebellum and some of its mechanisms. Exp Brain Res Suppl 6: 8-49. doi: 10.1007/978-3-642-68560-6_2
|
| [54] |
Burgoyne RD, Graham ME, Cambray-Deakin M (1993) Neurotrophic effects of NMDA receptor activation on developing cerebellar granule cells. J Neurocytol 22: 689-695. doi: 10.1007/BF01181314
|
| [55] |
Monti B, Contestabile A (2000) Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur J Neurosci 12: 3117-3123. doi: 10.1046/j.1460-9568.2000.00189.x
|
| [56] |
Hack N, Hidaka H, Wakefield MJ, et al. (1993) Promotion of granule cell survival by high K+ or excitatory amino acid treatment and Ca2+/calmodulin-dependent protein kinase activity. Neuroscience 57: 9-20. doi: 10.1016/0306-4522(93)90108-R
|
| [57] |
See V, Boutillier AR, Bito H, et al. (2001) Calcium/calmodulin-dependent protein kinase IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J 15: 134-144. doi: 10.1096/fj.00-0106com
|
| [58] |
Samhan-Arias AK, Martin-Romero FJ, Gutierrez-Merino C (2004) Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for the plasma membrane NADH oxidase activity in the commitment to apoptosis. Free Radic Biol Med 37: 48-61. doi: 10.1016/j.freeradbiomed.2004.04.002
|
| [59] | Schulz JB, Weller M, Klockgether T (1996) Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci 16: 4696-4706. |
| [60] |
Atlante A, Gagliardi S, Marra E, et al. (1998) Neuronal apoptosis in rats is accompanied by rapid impairment of cellular respiration and is prevented by scavengers of reactive oxygen species. Neurosci Lett 245: 127-130. doi: 10.1016/S0304-3940(98)00195-5
|
| [61] |
Hockenbery DM, Oltvai ZN, Yin X-M, et al. (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75: 241-251. doi: 10.1016/0092-8674(93)80066-N
|
| [62] |
Kane DJ, Sarafian TA, Anton R, et al. (1993) Bcl-2 inhibition of neuronal death: decreased generation of reactive oxygen species. Science 262: 1274-1277. doi: 10.1126/science.8235659
|
| [63] |
Mao GD, Poznansky MJ (1992) Electron spin resonance study on the permeability of superoxide radicals in lipid bilayers and biological membranes. FEBS Lett 305: 233-236. doi: 10.1016/0014-5793(92)80675-7
|
| [64] |
Samhan-Arias AK, Garcia-Bereguiain MA, Martin-Romero FJ, et al. (2009) Clustering of plasma membrane-bound cytochrome b5 reductase within ‘lipid rafts’ microdomains of the neuronal plasma membrane. Mol Cell Neurosci 40: 14-26. doi: 10.1016/j.mcn.2008.08.013
|
| [65] | Borgese N, Meldolesi J (1980) Localization and biosynthesis of NADH-cytochrome b5 reductase, an integral membrane protein, in rat liver cells. I. Distribution of the enzyme activity in microsomes, mitochondria, and Golgi complex. J Cell Biol 85: 501-515. |
| [66] |
Chatenay-Rivauday C, Cakar ZP, Jenö P, et al. (2004) Caveolae: biochemical analysis. Mol Biol Rep 31: 67-84. doi: 10.1023/B:MOLE.0000031352.51910.e9
|
| [67] | May JM (1999) Is ascorbic acid an antioxidant for the plasma membrane? FASEB J 13: 995-1006. |
| [68] |
Martin-Romero FJ, Gutierrez-Martin Y, Henao F, et al. (2002) The NADH oxidase activity of the plasma membrane of synaptosomes is a major source of superoxide anion and is inhibited by peroxynitrite. J Neurochem 82: 604-614. doi: 10.1046/j.1471-4159.2002.00983.x
|
| [69] |
Samhan-Arias AK, Duarte RO, Martin-Romero FJ, et al. (2008) Reduction of ascorbate free radical by the plasma membrane of synaptic terminals from rat brain. Arch Biochem Biophys 469: 243-254. doi: 10.1016/j.abb.2007.10.004
|
| [70] | Samhan-Arias AK, Gutierrez-Merino C (2014) Cytochrome b5 as a pleiotropic metabolic modulator in mammalian cells, In: Thom R. Editor, Cytochromes b and c: Biochemical properties, biological functions and electrochemical analysis, 1 Ed., New York (USA): Hauppauge, Chapter 2: 39-80. |
| [71] | Samhan-Arias AK, López-Sánchez C, Marques-da-Silva D, et al. (2015) High expression of cytochrome b5 reductase isoform 3/cytochrome b5 system in the cerebellum and pyramidal neurons of adult rat brain. Brain Struct Funct 1-16. |
| [72] | Percy MJ, Lappin TR (2008) Recessive congenital methaemoglobinaemia: cytochrome b5 reductase deficiency. Br J Haematol 141: 298-308. |
| [73] |
Ewenczyk C, Leroux A, Roubergue A, et al. (2008) Recessive hereditary methaemoglobinaemia, type II: delineation of the clinical spectrum. Brain 131: 760-761. doi: 10.1093/brain/awm337
|
| [74] |
Huang YH, Tai CL, Lu YH, et al. (2012) Recessive congenital methemoglobinemia caused by a rare mechanism: Maternal uniparental heterodisomy with segmental isodisomy of a chromosome 22. Blood Cells Mol Dis 49: 114-117. doi: 10.1016/j.bcmd.2012.05.005
|
| [75] |
Leroux A, Junien C, Kaplan J, et al. (1975) Generalised deficiency of cytochrome b5 reductase in congenital methaemoglobinaemia with mental retardation. Nature 258: 619-620. doi: 10.1038/258619a0
|
| [76] |
Toelle SP, Boltshauser E, Mössner E, et al. (2004) Severe neurological impairment in hereditary methaemoglobinaemia type 2. Eur J Pediatr 163: 207-209. doi: 10.1007/s00431-004-1409-x
|
| [77] | Aalfs CM, Salieb-Beugelaar GB, Wanders RJA, et al. (2000) A case of methemoglobinemia type II due to NADH-cytochrome b5 reductase deficiency: determination of the molecular basis. Hum Mutat 16: 18-22 |
| [78] | Marques-da-Silva D, Gutierrez-Merino C (2014) Caveolin-rich lipid rafts of the plasma membrane of mature cerebellar granule neurons are microcompartments for calcium/reactive oxygen and nitrogen species cross-talk signaling. Cell Calcium 56: 108-123. |
| [79] | Gutierrez-Merino C, Marques-da-Silva D, Fortalezas S, et al. (2014) Cytosolic calcium homeostasis in neurons: Control systems, modulation by reactive oxygen and nitrogen species, and space and time fluctuations, In: Heinbockel T. Editor, Neurochemistry, 1 Ed., Rijeka (Craotia): InTech, Chapter 3: 59-110. |
| [80] |
Pike LJ (2006) Rafts defined: a report on the keystone symposium on lipid rafts and cell function. J Lipid Res 47: 1597-1598. doi: 10.1194/jlr.E600002-JLR200
|
| [81] |
O’Connell KMM, Martens JR, Tamkun MM (2004) Localization of ion channels to lipid raft domains within the cardiovascular system. Trends Cardiovasc Med 14: 37-42. doi: 10.1016/j.tcm.2003.10.002
|
| [82] |
Head BP, Insel PA (2007) Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol 17: 51-57. doi: 10.1016/j.tcb.2006.11.008
|
| [83] | Wu G, Lu ZH, Nakamura K, et al. (1996) Trophic effect of cholera toxin B subunit in cultured cerebellar granule neurons: modulation of intracellular calcium by GM1 ganglioside. J Neurosci Res 44: 243-254. |
| [84] |
Wu G, Xie X, Lu ZH, et al. (2001) Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc Natl Acad Sci USA 98: 307-312. doi: 10.1073/pnas.98.1.307
|
| [85] |
Marques-da-Silva D, Samhan-Arias AK, Tiago T, et al. (2010) L-type calcium channels and cytochrome b5 reductase are components of protein complexes tightly associated with lipid rafts microdomains of the neuronal plasma membrane. J Proteomics 73: 1502-1510. doi: 10.1016/j.jprot.2010.02.014
|
| [86] |
Davare MA, Dong F, Rubin CS, et al. (1999) The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem 274: 30280-30287. doi: 10.1074/jbc.274.42.30280
|
| [87] |
Razani B, Rubin CS, Lisanti MP (1999) Regulation of cAMP-mediated Signal Transduction via Interaction of Caveolins with the Catalytic Subunit of Protein Kinase A. J Biol Chem 274: 26353-26360. doi: 10.1074/jbc.274.37.26353
|
| [88] | Suzuki T, Du F, Tian Q-B, et al. (2008) Ca2+/calmodulin-dependent protein kinase IIα clusters are associated with stable lipid rafts and their formation traps PSD-95. J Neurochem 104: 596-610. |
| [89] | Pinard CR, Mascagni F, McDonald AJ (2005) Neuronal localization of Cav1.2 L-type calcium channels in the rat basolateral amygdala. Brain Res 1064: 52 - 55. |
| [90] | Samhan-Arias AK, García-Bereguiaín MA, Gutierrez-Merino C (2007) Plasma membrane-bound cytochrome b5 reductase forms a large network of redox centres that co-localizes with cholera toxin B binding sites in cerebellar granule neurons in culture, In: Society for Free Radical Research (SFRR) Editor, Proceedings of the European Meeting of the SFFR, Bologna (Italy): Medimond, 147-150. |
| [91] | Samhan-Arias AK, Gutiérrez-Merino C (2008) Plasma membrane-bound cytochrome b5 reductase is associated with lipid rafts in cerebellar granule neurons in culture, In: Grune T. Editor, Proceedings of the European Meeting of the Society for Free Radical Research, 1 Ed., Bologna (Italy): Medimond, 75-78. |
| [92] | Silva DM, Samhan-Arias AK, Garcia-Bereguiain MA, et al. (2009) Major plasma membrane-associated redox centres co-localize with L-type calcium channels in neuronal lipid rafts microdomains, In: Caporosi D., Pigozzi F., Sabatini S. Editors, Free Radicals, Health and Lifestyle, 3 Eds., Bologna (Italy): Medimond, 127-130. |
| [93] |
Marques-da-Silva D, Gutierrez-Merino C (2012) L-type voltage-operated calcium channels, N-methyl-D-aspartate receptors and neuronal nitric-oxide synthase form a calcium/redox nano-transducer within lipid rafts. Biochem Biophys Res Commun 420: 257-262. doi: 10.1016/j.bbrc.2012.02.145
|
| [94] |
Sato Y, Sagami I, Shimizu T (2004) Identification of Caveolin-1-interacting Sites in Neuronal Nitric-oxide Synthase. J Biol Chem 279: 8827-8836. doi: 10.1074/jbc.M310327200
|
| [95] |
Samhan-Arias AK, Garcia-Bereguiain MA, Martin-Romero FJ, et al. (2006) Regionalization of plasma membrane-bound flavoproteins of cerebellar granule neurons in culture by fluorescence energy transfer imaging. J Fluorescence 16: 393-401. doi: 10.1007/s10895-005-0065-5
|
| [96] | Gutierrez-Merino C (2008) Redox modulation of neuronal calcium homeostasis and its deregulation by reactive oxygen species, In: Gutierrez-Merino C. and Leeuwenburgh C. Editors, Free Radicals in Biology and Medicine, 2 Eds., Kerala (India): Research Signpost, 67-101. |
| [97] |
Marchetti C, Usai C (1996) High affinity block by nimodipine of the internal calcium elevation in chronically depolarized rat cerebellar granule neurons. Neurosci Lett 207: 77-80. doi: 10.1016/0304-3940(96)12492-7
|
| [98] |
Maric D, Maric I, Barker JL (2000) Developmental changes in cell calcium homeostasis during neurogenesis of the embryonic rat cerebral cortex. Cereb Cortex 10: 561-573. doi: 10.1093/cercor/10.6.561
|
| [99] |
Arakawa Y, Nishijima C, Shimizu N, et al. (2002) Survival-promoting activity of nimodipine and nifedipine in rat motoneurons: implications of an intrinsic calcium toxicity in motoneurons. J Neurochem 83: 150-156. doi: 10.1046/j.1471-4159.2002.01126.x
|
| [100] |
Samhan-Arias AK, Gutierrez-Merino C (2014) Purified NADH-Cytochrome b5 Reductase Is a Novel Superoxide Anion Source Inhibited by Apocynin: Sensitivity to nitric oxide and peroxynitrite. Free Radic Biol Med 73: 174-189. doi: 10.1016/j.freeradbiomed.2014.04.033
|
| [101] |
Hidalgo C, Donoso P (2008) Crosstalk between calcium and redox signalling: from molecular mechanisms to health implications. Antioxid Redox Signal 10: 1275-1312. doi: 10.1089/ars.2007.1886
|
| [102] |
Szabó C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6: 662-680. doi: 10.1038/nrd2222
|
| [103] |
Bredt DS, Snyder SH (1994) Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem 63: 175-195. doi: 10.1146/annurev.bi.63.070194.001135
|
| [104] |
Parekh AB (2008) Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol 586: 3043-3054. doi: 10.1113/jphysiol.2008.153460
|
| [105] |
Neher E (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389-399. doi: 10.1016/S0896-6273(00)80983-6
|
| [106] | Neher E (1998) Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium 24: 345-357. |
| [107] |
Willmott NJ, Wong K, Strong AJ (2000) Intercellular Ca2+ waves in rat hippocampal slice and dissociated glial-neuron cultures mediated by nitric oxide. FEBS Lett 487: 239-247. doi: 10.1016/S0014-5793(00)02359-0
|
| [108] | Marques-da-Silva D (2012) Estudio de los microdominios de sistemas redox y de transporte de calcio en la membrana plasmática de neuronas. PhD Thesis, University of Extremadura. |
| [109] |
Contestabile A, Ciani E (2004) Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation. Neurochem Int 45: 903-914. doi: 10.1016/j.neuint.2004.03.021
|
| [110] |
Contestabile A (2008) Regulation of transcription factors by nitric oxide in neurons and in neural-derived tumor cells. Prog Neurobiol 84: 317-328. doi: 10.1016/j.pneurobio.2008.01.002
|
| [111] |
Grueter CE, Abiria SA, Wu Y, et al. (2008) Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel beta subunits. Biochemistry 47: 1760-1767. doi: 10.1021/bi701755q
|
| [112] |
Paratcha G, Ibáñez CF (2002) Lipid rafts and the control of neurotrophic factor signaling in the nervous system: variations on a theme. Curr Opin Neurobiol 12: 542-549. doi: 10.1016/S0959-4388(02)00363-X
|
| [113] | Inoue H, Miyaji M, Kosugi A, et al. (2002) Lipid rafts as the signaling scaffold for NK cell activation: tyrosine phosphorylation and association of LAT with phosphatidylinositol 3-kinase and phospholipase C-gamma following CD2 stimulation. Eur J Immunol 32: 2188-2198. |
| [114] |
Zheng F, Soellner D, Nunez J, et al. (2008) The basal level of intracellular calcium gates the activation of phosphoinositide 3-kinase - Akt signaling by brain-derived neurotrophic factor in cortical neurons. J Neurochem 106: 1259-1274. doi: 10.1111/j.1471-4159.2008.05478.x
|
| [115] |
Hudmon A, Schulman H, Kim J, et al. (2005) CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol 171: 537-547. doi: 10.1083/jcb.200505155
|
| [116] | Lee TS, Karl R, Moosmang S, et al. (2006) Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. J Biol Chem 281: 25560-25567. |
| [117] |
Coultrap SJ, Bayer KU (2014) Nitric Oxide Induces Ca2+-independent Activity of the Ca2+/Calmodulin-dependent Protein Kinase II (CaMKII). J Biol Chem 289: 19458-19465. doi: 10.1074/jbc.M114.558254
|
| [118] |
Coultrap SJ, Zaegel V, Bayer KU (2014) CaMKII isoforms differ in their specific requirements for regulation by nitric oxide. FEBS Lett 588: 4672-4676. doi: 10.1016/j.febslet.2014.10.039
|
| [119] | Müller U, Hildebrandt H (2002) Nitric Oxide/cGMP-Mediated Protein Kinase A Activation in the Antennal Lobes Plays an Important Role in Appetitive Reflex Habituation in the Honeybee. J Neurosci 22:8739-8747. |
| [120] |
De Jongh KS, Murphy BJ, Colvin AA, et al. (1996) Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3',5'-cyclic monophosphate-dependent protein kinase. Biochemistry 35: 10392-10340. doi: 10.1021/bi953023c
|
| [121] |
Mitterdorfer J, Froschmayr M, Grabner M, et al. (1996) Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel alpha 1 subunits. Biochemistry 35: 9400-9406. doi: 10.1021/bi960683o
|
| [122] |
Gao T, Yatani A, Dell’Acqua ML, et al. (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185-196. doi: 10.1016/S0896-6273(00)80358-X
|
| [123] |
Puri TS, Gerhardstein BL, Zhao XL, et al. (1997) Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry 36: 9605-9615. doi: 10.1021/bi970500d
|
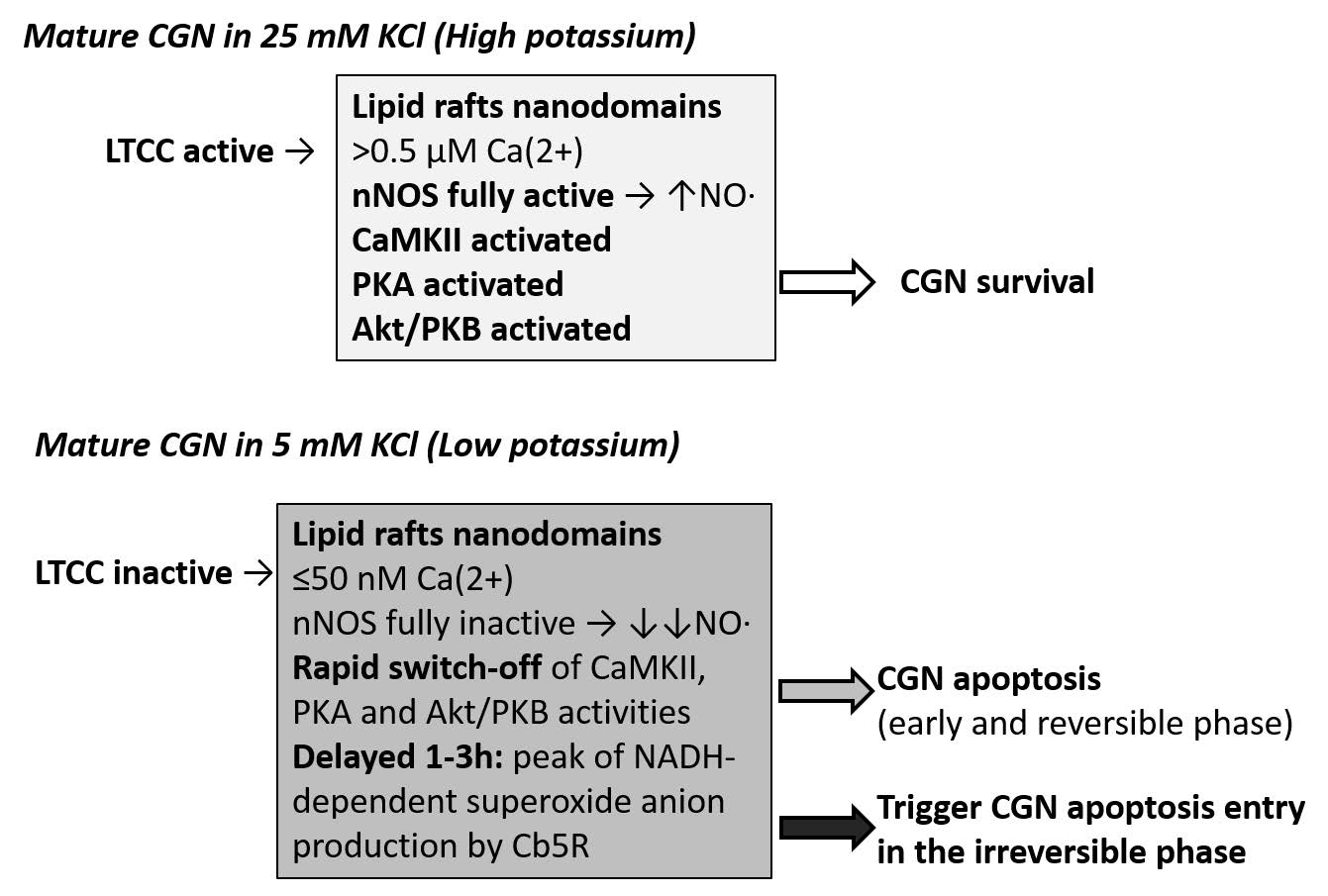
Figures(1)

Carlos Gutierrez-Merino, Dorinda Marques-da-Silva, Sofia Fortalezas, Alejandro K. Samhan-Arias. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracellular potassium deprivation[J]. AIMS Molecular Science, 2016, 3(1): 12-29. doi: 10.3934/molsci.2016.1.12







 DownLoad:
DownLoad: