Citation: Martin O. Steinhauser. Multiscale modeling, coarse-graining and shock wave computer simulations in materials science[J]. AIMS Materials Science, 2017, 4(6): 1319-1357. doi: 10.3934/matersci.2017.6.1319

| [1] | Phillips RR (2001) Crystals, defects and microstructures: Modeling across scales, Cambridge: Cambridge University Press. |
| [2] | Yip S (2005) Handbook of materials modeling, Berlin: Springer. |
| [3] | Steinhauser MO (2017) Computational Multiscale Modeling of Fluids and Solids-Theory and Applications, 2nd Edition, Berlin: Springer. |
| [4] |
McNeil PL, Terasaki M (2001) Coping with the inevitable: how cells repair a torn surface membrane. Nat Cell Biol 3: E124–E129. doi: 10.1038/35074652

|
| [5] | Schmidt M, Kahlert U, Wessolleck J, et al. (2014) Characterization of a setup to test the impact of high-amplitude pressure waves on living cells. Sci Rep 4: 3849. |
| [6] |
Gambihler S, Delius M, Ellwart JW (1992) Transient increase in membrane permeability of L1210 cells upon exposure to lithotripter shock waves in vitro. Naturwissenschaften 79: 328–329. doi: 10.1007/BF01138714
|
| [7] | Gambihler S, Delius M, Ellwart JW (1994) Permeabilization of the plasma membrane of L1210 mouse leukemia cells using lithotripter shock waves. J Membrane Biol 141: 267–275. |
| [8] | Kodama T, Doukas AG, Hamblin MR (2002) Shock wave-mediated molecular delivery into cells. BBA-Mol Cell Res 1542: 186–194. |
| [9] |
Bao G, Suresh S (2003) Cell and molecular mechanics of biological materials. Nat Mater 2: 715–725. doi: 10.1038/nmat1001
|
| [10] |
Tieleman DP, Leontiadou H, Mark AE, et al. (2003) Simulation of Pore Formation in Lipid Bilayers by Mechanical Stress and Electric Fields. J Am Chem Soc 125: 6382–6383. doi: 10.1021/ja029504i
|
| [11] |
Sundaram J, Mellein BR, Mitragotri S (2003) An Experimental and Theoretical Analysis of Ultrasound-Induced Permeabilization of Cell Membranes. Biophys J 84: 3087–3101. doi: 10.1016/S0006-3495(03)70034-4
|
| [12] |
Doukas AG, Kollias N (2004) Transdermal drug delivery with a pressure wave. Adv Drug Deliver Rev 56: 559–579. doi: 10.1016/j.addr.2003.10.031
|
| [13] |
Coussios CC, Roy RA (2008) Applications of Acoustics and Cavitation to Noninvasive Therapy and Drug Delivery. Annu Rev Fluid Mech 40: 395–420. doi: 10.1146/annurev.fluid.40.111406.102116
|
| [14] |
Prausnitz MR, Langer R (2008) Transdermal drug delivery. Nat Biotechnol 26: 1261–1268. doi: 10.1038/nbt.1504
|
| [15] |
Ashley CE, Carnes EC, Phillips GK, et al. (2011) The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater 10: 389–397. doi: 10.1038/nmat2992
|
| [16] |
Koshiyama K, Wada S (2011) Molecular dynamics simulations of pore formation dynamics during the rupture process of a phospholipid bilayer caused by high-speed equibiaxial stretching. J Biomech 44: 2053–2058. doi: 10.1016/j.jbiomech.2011.05.014
|
| [17] |
Steinhauser MO (2016) On the Destruction of Cancer Cells Using Laser-Induced Shock-Waves: A Review on Experiments and Multiscale Computer Simulations. Radiol Open J 1: 60–75. doi: 10.17140/ROJ-1-110
|
| [18] | Krehl POK (2009) History of Shock Waves, Explosions and Impact: A Chronological and Biographical Reference, Berlin: Springer. |
| [19] |
Steinhauser MO, Schneider J, Blumen A (2009) Simulating dynamic crossover behavior of semiflexible linear polymers in solution and in the melt. J Chem Phys 130: 164902. doi: 10.1063/1.3111038
|
| [20] |
Rodriguez V, Saurel R, Jourdan G, et al. (2013) Solid-particle jet formation under shock-wave acceleration. Phys Rev E 88: 063011. doi: 10.1103/PhysRevE.88.063011
|
| [21] |
Zheng J, Chen QF, Gu YJ, et al. (2012) Hugoniot measurements of double-shocked precompressed dense xenon plasmas. Phys Rev E 86: 066406. doi: 10.1103/PhysRevE.86.066406
|
| [22] |
Falk K, Regan SP, Vorberger J, et al. (2013) Comparison between x-ray scattering and velocityinterferometry measurements from shocked liquid deuterium. Phys Rev E 87: 043112. doi: 10.1103/PhysRevE.87.043112
|
| [23] |
Brujan EA, Matsumoto Y (2014) Shock wave emission from a hemispherical cloud of bubbles in non-Newtonian fluids. J Non-Newton Fluid 204: 32–37. doi: 10.1016/j.jnnfm.2013.12.003
|
| [24] |
Iakovlev S, Iakovlev S, Buchner C, et al. (2014) Resonance-like phenomena in a submerged cylindrical shell subjected to two consecutive shock waves: The effect of the inner fluid. J Fluid Struct 50: 153–170. doi: 10.1016/j.jfluidstructs.2014.05.013
|
| [25] |
Bringa EM, Caro A,Wang YM, et al. (2005) Ultrahigh strength in nanocrystalline materials under shock loading. Science 309: 1838–1841. doi: 10.1126/science.1116723
|
| [26] |
Kadau K, Germann TC, Lomdahl PS, et al. (2007) Shock waves in polycrystalline iron. Phys Rev Lett 98: 135701. doi: 10.1103/PhysRevLett.98.135701
|
| [27] |
Knudson MD, Desjarlais MP, Dolan DH (2008) Shock-Wave Exploration of the High-Pressure Phases of Carbon. Science 322: 1822–1825. doi: 10.1126/science.1165278
|
| [28] |
Gurnett DA, Kurth WS (2005) Electron plasma oscillations upstream of the solar wind termination shock. Science 309: 2025–2027. doi: 10.1126/science.1117425
|
| [29] |
Gurnett DA, Kurth WS (2008) Intense plasma waves at and near the solar wind termination shock. Nature 454: 78–80. doi: 10.1038/nature07023
|
| [30] |
Dutton Z, Budde M, Slowe C, et al. (2001) Observation of quantum shock waves created with ultra-compressed slow light pulses in a Bose-Einstein condensate. Science 293: 663–668. doi: 10.1126/science.1062527
|
| [31] |
Damski B (2006) Shock waves in a one-dimensional Bose gas: From a Bose-Einstein condensate to a Tonks gas. Phys Rev A 73: 043601. doi: 10.1103/PhysRevA.73.043601
|
| [32] |
Chang JJ, Engels P, Hoefer MA (2008) Formation of dispersive shock waves by merging and splitting Bose-Einstein condensates. Phys Rev Lett 101: 170404. doi: 10.1103/PhysRevLett.101.170404
|
| [33] | Millot M, Dubrovinskaia N, Černok A, et al. (2015) Planetary science. Shock compression of stishovite and melting of silica at planetary interior conditions. Science 347: 418–420. |
| [34] |
Bridge HS, Lazarus AJ, Snyder CW, et al. (1967) Mariner V: Plasma and Magnetic Fields Observed near Venus. Science 158: 1669–1673. doi: 10.1126/science.158.3809.1669
|
| [35] |
McKee CF, Draine BT (1991) Interstellar shock waves. Science 252: 397–403. doi: 10.1126/science.252.5004.397
|
| [36] | McClure S, Dorfmüller C (2002) Extracorporeal shock wave therapy: Theory and equipment. Clin Tech Equine Pract 2: 348–357. |
| [37] |
Lingeman JE, McAteer JA, Gnessin E, et al. (2009) Shock wave lithotripsy: advances in technology and technique. Nat Rev Urol 6: 660–670. doi: 10.1038/nrurol.2009.216
|
| [38] | Cherenkov PA (1934) Visible emission of clean liquids by action of gamma radiation. Dokl Akad Nauk SSSR 2: 451–454. |
| [39] |
Mach E, Salcher P (1887) Photographische Fixirung der durch Projectile in der Luft eingeleiteten Vorgänge. Ann Phys 268: 277–291. doi: 10.1002/andp.18872681008
|
| [40] |
Kühn M, Steinhauser MO (2008) Modeling and simulation of microstructures using power diagrams: Proof of the concept. Appl Phys Lett 93: 034102. doi: 10.1063/1.2959733
|
| [41] |
Walsh JM, Rice MH (1957) Dynamic compression of liquids from measurements on strong shock waves. J Chem Phys 26: 815–823. doi: 10.1063/1.1743414
|
| [42] | Asay JR, Chhabildas LC (2003) Paradigms and Challenges in Shock Wave Research, High-Pressure Shock Compression of Solids VI, New York: Springer-Verlag New York, 57–119. |
| [43] |
Steinhauser MO, Grass K, Strassburger E, et al. (2009) Impact failure of granular materials-Nonequilibrium multiscale simulations and high-speed experiments. Int J Plasticity 25: 161–182. doi: 10.1016/j.ijplas.2007.11.002
|
| [44] |
Watson E, Steinhauser MO (2017) Discrete Particle Method for Simulating Hypervelocity Impact Phenomena. Materials 10: 379. doi: 10.3390/ma10040379
|
| [45] |
Holian BL, Lomdahl PS (1998) Plasticity induced by shock waves in nonequilibrium moleculardynamics simulations. Science 280: 2085–2088. doi: 10.1126/science.280.5372.2085
|
| [46] |
Kadau K, Germann TC, Lomdahl PS, et al. (2002) Microscopic view of structural phase transitions induced by shock waves. Science 296: 1681–1684. doi: 10.1126/science.1070375
|
| [47] |
Chen M, McCauley JW, Hemker KJ (2003) Shock-Induced Localized Amorphization in Boron Carbide. Science 299: 1563–1566. doi: 10.1126/science.1080819
|
| [48] | Holian BL (2004) Molecular dynamics comes of age for shockwave research. Shock Waves 13: 489–495. |
| [49] |
Germann TC, Kadau K (2008) Trillion-atom molecular dynamics becomes a reality. Int J Mod Phys C 19: 1315–1319. doi: 10.1142/S0129183108012911
|
| [50] | Ciccotti G, Frenkel G, McDonald IR (1987) Simulation of Liquids and Solids, Amsterdam: North-Holland. |
| [51] | Allen MP, Tildesley DJ (1987) Computer Simulation of Liquids, Oxford, UK: Clarendon Press. |
| [52] |
Liu WK, Hao S, Belytschko T, et al. (1999) Multiple scale meshfree methods for damage fracture and localization. Comp Mater Sci 16: 197–205. doi: 10.1016/S0927-0256(99)00062-2
|
| [53] |
Gates TS, Odegard GM, Frankland SJV, et al. (2005) Computational materials: Multi-scale modeling and simulation of nanostructured materials. Compos Sci Technol 65: 2416–2434. doi: 10.1016/j.compscitech.2005.06.009
|
| [54] | Steinhauser MO (2013) Computer Simulation in Physics and Engineering, 1st Edition, Berlin: deGruyter. |
| [55] |
Finnis MW, Sinclair JE (1984) A simple empirical N-body potential for transition metals. Philos Mag A 50: 45–55. doi: 10.1080/01418618408244210
|
| [56] |
Kohn W (1996) Density functional and density matrix method scaling linearly with the number of atoms. Phys Rev Lett 76: 3168–3171. doi: 10.1103/PhysRevLett.76.3168
|
| [57] |
Car R, Parrinello M (1985) Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55: 2471–2474. doi: 10.1103/PhysRevLett.55.2471
|
| [58] |
Elstner M, Porezag D, Jungnickel G, et al. (1998) Self-consistent-charge density-functional tightbinding method for simulations of complex materials properties. Phys Rev B 58: 7260–7268. doi: 10.1103/PhysRevB.58.7260
|
| [59] |
Sutton AP, Finnis MW, Pettifor DG, et al. (1988) The tight-binding bond model. J Phys C-Solid State Phys 21: 35–66. doi: 10.1088/0022-3719/21/1/007
|
| [60] | Szabo A, Ostlund NS (1996) Modern quantum chemistry: introduction to advanced electronic structure theory, (Dover Books on Chemistry), New York: Dover Publications. |
| [61] |
Kadau K, Germann TC, Lomdahl PS (2006) Molecular dynamics comes of age: 320 billion atom simulation on BlueGene/L. Int J Mod Phys C 17: 1755–1761. doi: 10.1142/S0129183106010182
|
| [62] |
Fineberg J (2003) Materials science: close-up on cracks. Nature 426: 131–132. doi: 10.1038/426131a
|
| [63] |
Buehler M, Hartmaier A, Gao H, et al. (2004) Atomic plasticity: description and analysis of a onebillion atom simulation of ductile materials failure. Comput Method Appl M 193: 5257–5282. doi: 10.1016/j.cma.2003.12.066
|
| [64] |
Abraham FF, Gao HJ (2000) How fast can cracks propagate? Phys Rev Lett 84: 3113–3116. doi: 10.1103/PhysRevLett.84.3113
|
| [65] |
Bulatov V, Abraham FF, Kubin L, et al. (1998) Connecting atomistic and mesoscale simulations of crystal plasticity. Nature 391: 669–672. doi: 10.1038/35577
|
| [66] |
Gross SP, Fineberg J, Marder M, et al. (1993) Acoustic emissions from rapidly moving cracks. Phys Rev Lett 71: 3162–3165. doi: 10.1103/PhysRevLett.71.3162
|
| [67] | Courant R (1943) Variational Methods for the Solution of Problems of Equilibrium and Vibrations. B Am Math Soc 49: 1–23. |
| [68] |
Lucy LB (1977) A numerical approach to the testing of the fission hypothesis. Astron J 82: 1013–1024. doi: 10.1086/112164
|
| [69] |
Cabibbo N, Iwasaki Y, Schilling K (1999) High performance computing in lattice QCD. Parallel Comput 25: 1197–1198. doi: 10.1016/S0167-8191(99)00045-9
|
| [70] |
Evertz HG (2003) The loop algorithm. Adv Phys 52: 1–66. doi: 10.1080/0001873021000049195
|
| [71] | Holm EA, Battaile CC (2001) The computer simulation of microstructural evolution. JOM 53: 20–23. |
| [72] |
Nielsen SO, Lopez CF, Srinivas G, et al. (2004) Coarse grain models and the computer simulation of soft materials. J Phys-Condens Mat 16: 481–512. doi: 10.1088/0953-8984/16/15/R03
|
| [73] |
Praprotnik M, Site LD, Kremer K (2008) Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu Rev Phys Chem 59: 545–571. doi: 10.1146/annurev.physchem.59.032607.093707
|
| [74] | Karimi-Varzaneh HA, Müller-Plathe F (2011) Coarse-Grained Modeling for Macromolecular Chemistry, In: Kirchner B, Vrabec J, Topics in Current Chemistry, Berlin, Heidelberg: Springer, 326–321. |
| [75] | Müller-Plathe F (2002) Coarse-graining in polymer simulation: from the atomistic to the mesoscopic scale and back. Chem Phys Chem 3: 755–769. |
| [76] |
Abraham FF, Broughton JQ, Broughton JQ, et al. (1998) Spanning the length scales in dynamic simulation. Comp Phys 12: 538–546. doi: 10.1063/1.168756
|
| [77] |
Abraham FF, Brodbeck D, Rafey R, et al. (1994) Instability dynamics of fracture: A computer simulation investigation. Phys Rev Lett 73: 272–275. doi: 10.1103/PhysRevLett.73.272
|
| [78] |
Abraham FF, Brodbeck D, Rudge WE, et al. (1998) Ab initio dynamics of rapid fracture. Model Simul Mater Sc 6: 639–670. doi: 10.1088/0965-0393/6/5/010
|
| [79] | Warshel A, LevittM(1976) Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol 103: 227–249. |
| [80] | Winkler RG, Steinhauser MO, Reineker P (2002) Complex formation in systems of oppositely charged polyelectrolytes: a molecular dynamics simulation study. Phys Rev E 66: 021802. |
| [81] |
Dünweg B, Reith D, Steinhauser M, et al. (2002) Corrections to scaling in the hydrodynamic properties of dilute polymer solutions. J Chem Phys 117: 914–924. doi: 10.1063/1.1483296
|
| [82] |
Stevens MJ (2004) Coarse-grained simulations of lipid bilayers. J Chem Phys 121: 11942–11948. doi: 10.1063/1.1814058
|
| [83] | Steinhauser MO (2005) A molecular dynamics study on universal properties of polymer chains in different solvent qualities. Part I. A review of linear chain properties. J Chem Phys 122: 094901. |
| [84] |
Steinhauser MO, Hiermaier S (2009) A Review of Computational Methods in Materials Science: Examples from Shock-Wave and Polymer Physics. Int J Mol Sci 10: 5135–5216. doi: 10.3390/ijms10125135
|
| [85] |
Goetz R, Gompper G, Lipowsky R (1999) Mobility and elasticity of self-assembled membranes. Phys Rev Lett 82: 221–224. doi: 10.1103/PhysRevLett.82.221
|
| [86] |
Lipowsky R (2004) Biomimetic membrane modelling: pictures from the twilight zone. Nat Mater 3: 589–591. doi: 10.1038/nmat1208
|
| [87] |
Lyubartsev AP (2005) Multiscale modeling of lipids and lipid bilayers. Eur Biophys J 35: 53–61. doi: 10.1007/s00249-005-0005-y
|
| [88] |
Orsi M, Michel J, Essex JW (2010) Coarse-grain modelling of DMPC and DOPC lipid bilayers. J Phys-Condens Mat 22: 155106. doi: 10.1088/0953-8984/22/15/155106
|
| [89] | Steinhauser MO (2012) Introduction to Molecular Dynamics Simulations: Applications in Hard and Soft Condensed Matter Physics, InTech. |
| [90] | Alberts B, Bray D, Johnson A, et al. (2000) Molecular Biology of the Cell, 4 Edition, New York: Garland Science, Taylor and Francis Group. |
| [91] |
Steinhauser MO, Steinhauser MO, Schmidt M (2014) Destruction of cancer cells by laserinduced shock waves: recent developments in experimental treatments and multiscale computer simulations. Soft Matter 10: 4778–4788. doi: 10.1039/C4SM00407H
|
| [92] | Tozzini V (2004) Coarse-grained models for proteins. Curr Opin Struc Biol 15: 144–150. |
| [93] |
Ayton GS, Noid WG, Voth GA (2007) Multiscale modeling of biomolecular systems: in serial and in parallel. Curr Opin Struc Biol 17: 192–198. doi: 10.1016/j.sbi.2007.03.004
|
| [94] |
Forrest LR, Sansom MS (2000) Membrane simulations: bigger and better? Curr Opin Struc Biol 10: 174–181. doi: 10.1016/S0959-440X(00)00066-X
|
| [95] |
Woods CJ, Mulholland AJ (2008) Multiscale modelling of biological systems. RSC Special Periodicals Report: Chemical Modelling, Applications and Theory 5: 13–50. doi: 10.1039/b608778g
|
| [96] | Steinhauser MO (editor) (2016) Special Issue of the Journal Materials: Computational Multiscale Modeling and Simulation in Materials Science. Available from: http://www.mdpi.com/journal/materials/special issues/modeling and simulation. |
| [97] |
Brendel W (1986) Shock Waves: A New Physical Principle in Medicine. Eur Surg Res 18: 177–180. doi: 10.1159/000128523
|
| [98] | Wang CJ (2003) An overview of shock wave therapy in musculoskeletal disorders. Chang Gung Med J 26: 220–232. |
| [99] |
Wang ZJZ, DesernoM (2010) A systematically coarse-grained solvent-free model for quantitative phospholipid bilayer simulations. J Phys Chem B 114: 11207–11220. doi: 10.1021/jp102543j
|
| [100] |
Wang ZB, Wu J, Fang LQ, et al. (2011) Preliminary ex vivo feasibility study on targeted cell surgery by high intensity focused ultrasound (HIFU). Ultrasonics 51: 369–375. doi: 10.1016/j.ultras.2010.11.002
|
| [101] |
Wang S, Frenkel V, Zderic V (2011) Optimization of pulsed focused ultrasound exposures for hyperthermia applications. J Acoust Soc Am 130: 599–609. doi: 10.1121/1.3598464
|
| [102] |
Paul W, Smith GD, Yoon DY (1997) Static and dynamic properties of an-C100H202 melt from molecular dynamics simulations. Macromolecules 30: 7772–7780. doi: 10.1021/ma971184d
|
| [103] |
Kreer T, Baschnagel J, Mueller M, et al. (2001) Monte Carlo Simulation of long chain polymer melts: Crossover from Rouse to reptation dynamics. Macromolecules 34: 1105–1117. doi: 10.1021/ma001500f
|
| [104] |
Krushev S, Paul W, Smith GD (2002) The role of internal rotational barriers in polymer melt chain dynamics. Macromolecules 35: 4198–4203. doi: 10.1021/ma0115794
|
| [105] |
Bulacu M, van der Giessen E (2005) Effect of bending and torsion rigidity on self-diffusion in polymer melts: A molecular-dynamics study. J Chem Phys 123: 114901. doi: 10.1063/1.2035086
|
| [106] | Kratky O, Porod G (1949) Röntgenuntersuchung gelöster Fadenmoleküle. Recl Trva Chim Pays-Bas 68: 1106–1122. |
| [107] | Doi M, Edwards SF (1986) The Theory of Polymer Dynamics, Oxford: Clarendon Press. |
| [108] |
Harris RA, Hearst JE (1966) On Polymer Dynamics. J Chem Phys 44: 2595–2602. doi: 10.1063/1.1727098
|
| [109] | Hearst JE, Harris RA (1967) On Polymer Dynamics. III. Elastic Light Scattering. J Chem Phys 46: 398–398. |
| [110] |
Harnau L, Winkler RG, Reineker P (1997) Influence of stiffness on the dynamics of macromolecules in a melt. J Chem Phys 106: 2469–2476. doi: 10.1063/1.473154
|
| [111] |
Harnau L, WInkler RG, Reineker P (1999) On the dynamics of polymer melts: Contribution of Rouse and bending modes. EPL 45: 488–494. doi: 10.1209/epl/i1999-00193-6
|
| [112] |
Steinhauser MO (2008) Static and dynamic scaling of semiflexible polymer chains-a molecular dynamics simulation study of single chains and melts. Mech Time-Depend Mat 12: 291–312. doi: 10.1007/s11043-008-9062-9
|
| [113] |
Guenza M (2003) Cooperative dynamics in semiflexibile unentangled polymer fluids. J Chem Phys 119: 7568–7578. doi: 10.1063/1.1606674
|
| [114] | Piran T (2004) Statistical Mechanics of Membranes and Interfaces, 2 edition, World Scientific Publishing Co., Inc. |
| [115] |
Schindler T, Kröner D, Steinhauser MO (2016) On the dynamics of molecular self-assembly and the structural analysis of bilayer membranes using coarse-grained molecular dynamics simulations. BBA-Biomembranes 1858: 1955–1963. doi: 10.1016/j.bbamem.2016.05.014
|
| [116] |
Brannigan G, Lin LCL, Brown FLH (2006) Implicit solvent simulation models for biomembranes. Eur Biophys J 35: 104–124. doi: 10.1007/s00249-005-0013-y
|
| [117] |
Chang R, Ayton GS, Voth GA (2005) Multiscale coupling of mesoscopic- and atomistic-level lipid bilayer simulations. J Chem Phys 122: 244716. doi: 10.1063/1.1931651
|
| [118] |
Huang MJ, Kapral R, Mikhailov AS, et al. (2012) Coarse-grain model for lipid bilayer selfassembly and dynamics: Multiparticle collision description of the solvent. J Chem Phys 137: 055101. doi: 10.1063/1.4736414
|
| [119] |
Pandit SA, Scott HL (2009) Multiscale simulations of heterogeneous model membranes. BBA-Biomembranes 1788: 136–148. doi: 10.1016/j.bbamem.2008.09.004
|
| [120] |
Farago O (2003) "Water-free" computer model for fluid bilayer membranes. J Chem Phys 119: 596–605. doi: 10.1063/1.1578612
|
| [121] |
Brannigan G, Philips PF, Brown FLH (2005) Flexible lipid bilayers in implicit solvent. Phys Rev E 72: 011915. doi: 10.1103/PhysRevE.72.011915
|
| [122] |
Yuan H, Huang C, Li J, et al. (2010) One-particle-thick, solvent-free, coarse-grained model for biological and biomimetic fluid membranes. Phys Rev E 82: 011905. doi: 10.1103/PhysRevE.82.011905
|
| [123] |
Noguchi H (2011) Solvent-free coarse-grained lipid model for large-scale simulations. J Chem Phys 134: 055101. doi: 10.1063/1.3541246
|
| [124] |
Weiner SJ, Kollman PA, Case DA, et al. (1984) A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 106: 765–784. doi: 10.1021/ja00315a051
|
| [125] |
Paul W, Yoon DY, Smith GD, et al. (1995) An Optimized United Atom Model for Simulations of Polymethylene Melts. J Chem Phys 103: 1702–1709. doi: 10.1063/1.469740
|
| [126] |
Siu SWI, Vácha R, Jungwirth P, et al. (2008) Biomolecular simulations of membranes: physical properties from different force fields. J Phys Chem 128: 125103. doi: 10.1063/1.2897760
|
| [127] |
Drouffe JM, Maggs AC, Leibler S, et al. (1991) Computer simulations of self-assembled membranes. Science 254: 1353–1356. doi: 10.1126/science.1962193
|
| [128] |
Goetz R, Lipowsky R (1998) Computer simulations of bilayer membranes: Self-assembly and interfacial tension. J Chem Phys 108: 7397–7409. doi: 10.1063/1.476160
|
| [129] |
Noguchi H, Takasu M (2001) Self-assembly of amphiphiles into vesicles: A Brownian dynamics simulation. Phys Rev E 64: 041913. doi: 10.1103/PhysRevE.64.041913
|
| [130] |
Bourov GK, Bhattacharya A (2005) Brownian dynamics simulation study of self-assembly of amphiphiles with large hydrophilic heads. J Chem Phys 122: 44702. doi: 10.1063/1.1834495
|
| [131] |
Steinhauser MO, Grass K, Thoma K, et al. (2006) Impact dynamics and failure of brittle solid states by means of nonequilibrium molecular dynamics simulations. EPL 73: 62–68. doi: 10.1209/epl/i2005-10353-2
|
| [132] |
Yang S, Qu J (2014) Coarse-grained molecular dynamics simulations of the tensile behavior of a thermosetting polymer. Phys Rev E 90: 012601. doi: 10.1103/PhysRevE.90.012601
|
| [133] | Eslami H, Müller-Plathe F (2013) How thick is the interphase in an ultrathin polymer film? Coarse-grained molecular dynamics simulations of polyamide-6,6 on graphene. J Phys Chem 117: 5249–5257. |
| [134] |
Ganzenm¨uller GC, Hiermaier S, Steinhauser MO (2011) Shock-wave induced damage in lipid bilayers: a dissipative particle dynamics simulation study. Soft Matter 7: 4307–4317. doi: 10.1039/c0sm01296c
|
| [135] |
Huang WX, Chang CB, Sung HJ (2012) Three-dimensional simulation of elastic capsules in shear flow by the penalty immersed boundary method. J Comput Phys 231: 3340–3364. doi: 10.1016/j.jcp.2012.01.006
|
| [136] | Pazzona FG, Demontis P (2012) A grand-canonical Monte Carlo study of the adsorption properties of argon confined in ZIF-8: local thermodynamic modeling. J Phys Chem 117: 349–357. |
| [137] |
Pogodin S, Baulin VA (2010) Coarse-grained models of phospholipid membranes within the single chain mean field theory. Soft Matter 6: 2216–2226. doi: 10.1039/b927437e
|
| [138] |
Wang Y, Sigurdsson JK, Brandt E, et al. (2013) Dynamic implicit-solvent coarse-grained models of lipid bilayer membranes: fluctuating hydrodynamics thermostat. Phys Rev E 88: 023301. doi: 10.1103/PhysRevE.88.023301
|
| [139] |
Koshiyama K, Kodama T, Yano T, et al. (2006) Structural Change in Lipid Bilayers and Water Penetration Induced by Shock Waves: Molecular Dynamics Simulations. Biophys J 91: 2198–2205. doi: 10.1529/biophysj.105.077677
|
| [140] |
Koshiyama K, Kodama T, Yano T, et al. (2008) Molecular dynamics simulation of structural changes of lipid bilayers induced by shock waves: Effects of incident angles. BBA-Biomembranes 1778: 1423–1428. doi: 10.1016/j.bbamem.2008.03.010
|
| [141] |
Lechuga J, Drikakis D, Pal S (2008) Molecular dynamics study of the interaction of a shock wave with a biological membrane. Int J Numer Mech Fluids 57: 677–692. doi: 10.1002/fld.1588
|
| [142] |
Kodama T, Kodama T, Hamblin MR, et al. (2000) Cytoplasmic molecular delivery with shock waves: importance of impulse. Biophys J 79: 1821–1832. doi: 10.1016/S0006-3495(00)76432-0
|
| [143] |
Doukas AG, McAuliffe DJ, Lee S, et al. (1995) Physical factors involved in stress-wave-induced cell injury: The effect of stress gradient. Ultrasound Med Biol 21: 961–967. doi: 10.1016/0301-5629(95)00027-O
|
| [144] |
Doukas AG, Flotte TJ (1996) Physical characteristics and biological effects of laser-induced stress waves. Ultrasound Med Biol 22: 151–164. doi: 10.1016/0301-5629(95)02026-8
|
| [145] |
Lee S, Doukas AG (1999) Laser-generated stress waves and their effects on the cell membrane. IEEE J Sel Top Quant 5: 997–1003. doi: 10.1109/2944.796322
|
| [146] |
Español P (1997) Dissipative Particle Dynamics with energy conservation. EPL 40: 631–636. doi: 10.1209/epl/i1997-00515-8
|
| [147] |
Steinhauser MO, Schindler T (2017) Particle-based simulations of bilayer membranes: selfassembly, structural analysis, and shock-wave damage. Comp Part Mech 4: 69–86. doi: 10.1007/s40571-016-0126-3
|
| [148] | Hansen JP, McDonald IR (2005) Theory of Simple Liquids, Academic Press. |

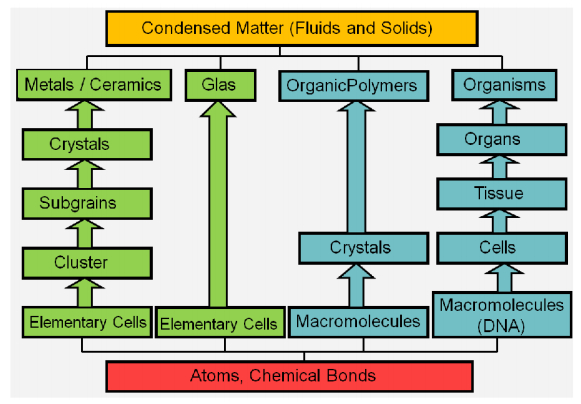
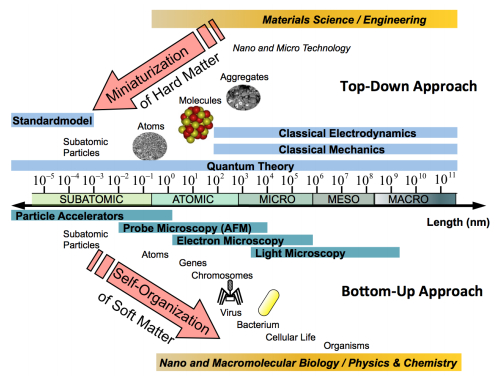
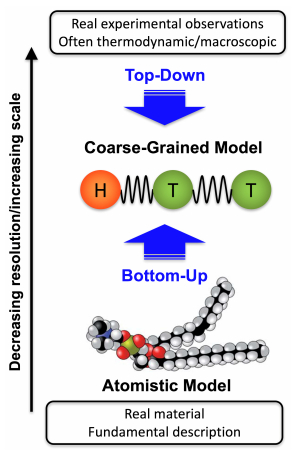
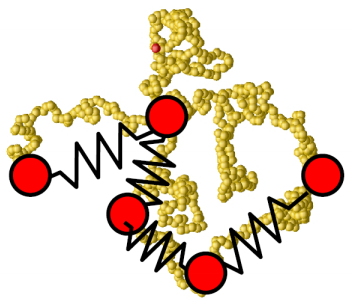
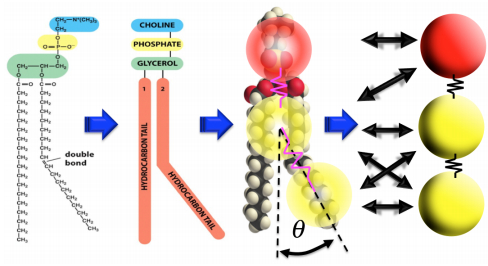
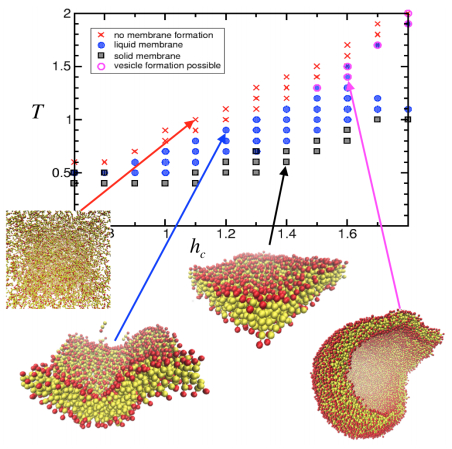
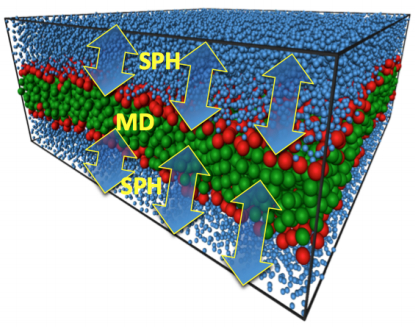
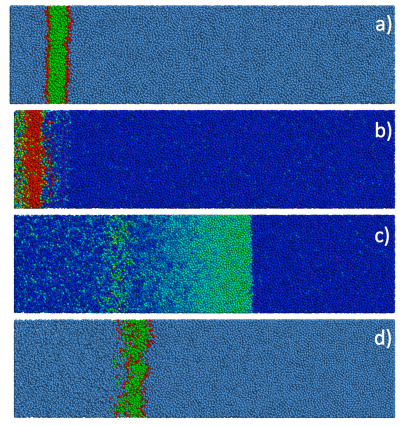
Figures(22)

Martin O. Steinhauser. Multiscale modeling, coarse-graining and shock wave computer simulations in materials science[J]. AIMS Materials Science, 2017, 4(6): 1319-1357. doi: 10.3934/matersci.2017.6.1319







 DownLoad:
DownLoad: