Citation: M.Bansbach Heather, H.Guilford William. Actin nitrosylation and its effect on myosin driven motility[J]. AIMS Molecular Science, 2016, 3(3): 426-438. doi: 10.3934/molsci.2016.3.426

| [1] | Kobzik L, Reid MB, Bredt DS, et al. (1994) Nitric oxide in skeletal muscle. Nature 372: 546-548. |
| [2] | Stamler JS, Meissner G (2001) Physiology of nitric oxide in skeletal muscle. Physiol Rev 81: 209-237. |
| [3] | Andrade FH, Reid MB, Allen DG, et al. (1998) Effect of nitric oxide on single skeletal muscle fibres from the mouse. J Physiol 509: 577-586. |
| [4] | Galler S, Hilber K, Göbesberger A (1997) Effects of nitric oxide on force-generating proteins of skeletal muscle. Pflüg Arch Eur J Physiol 434: 242-245. |
| [5] | Perkins WJ, Han YS, Sieck GC (1997) Skeletal muscle force and actomyosin ATPase activity reduced by nitric oxide donor. J Appl Physiol 83: 1326-1332. |
| [6] | Takahashi S, Shoji H, Okabe E (1999) Intracellular effects of nitric oxide on cardiac myofibrillar function. Free Radic Biol Med 27: S87. |
| [7] | Evangelista AM, Rao VS, Filo AR, et al. (2010) Direct Regulation of Striated Muscle Myosins by Nitric Oxide and Endogenous Nitrosothiols. PLoS One 5: e11209. |
| [8] | Lu J, Katano T, Uta D, et al. (2011) Rapid S-nitrosylation of actin by NO-generating donors and in inflammatory pain model mice. Mol Pain 7: 101. |
| [9] | Rubenstein PA (1990) The functional importance of multiple actin isoforms. Bioessays News Rev Mol Cell Dev Biol 12: 309-315. |
| [10] | Khaitlina SY (2001) Functional specificity of actin isoforms. Int Rev Cytol 202: 35-98. |
| [11] | Elzinga M, Collins JH, Kuehl WM, et al. (1973) Complete Amino-Acid Sequence of Actin of Rabbit Skeletal Muscle. Proc Natl Acad Sci U S A 70: 2687-2691. |
| [12] | Bizzozero OA, Zheng J (2009) Identification of major S‐nitrosylated proteins in murine experimental autoimmune encephalomyelitis, Identification of major S‐nitrosylated proteins in murine experimental autoimmune encephalomyelitis, J Neurosci Res 87: 2881-2889. |
| [13] | Chen SC, Huang B, Liu YC, et al. (2008) Acute hypoxia enhances proteins’ S-nitrosylation in endothelial cells. Biochem Biophys Res Commun 377: 1274-1278. |
| [14] | Dalle-Donne I, Milzani A, Giustarini D, et al. (2000) S-NO-actin: S-nitrosylation kinetics and the effect on isolated vascular smooth muscle. J Muscle Res Cell Motil 21: 171-181. |
| [15] | Guo B, Guilford WH (2004) The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motil Cytoskeleton 59: 264-272. |
| [16] | Rao VS, Marongelli EN, Guilford WH (2009) Phosphorylation of tropomyosin extends cooperative binding of myosin beyond a single regulatory unit. Cell Motil Cytoskeleton 66: 10-23. |
| [17] | Miller-Jaster KN, Petrie Aronin CE, Guilford WH (2011) A Quantitative Comparison of Blocking Agents in the In Vitro Motility Assay. Cell Mol Bioeng 5: 44-51. |
| [18] | Han P, Zhou X, Huang B, et al. (2008) On-gel fluorescent visualization and the site identification of S-nitrosylated proteins. Anal Biochem 377: 150-155. |
| [19] | Rasband WS, ImageJ. National Institutes of Health, 1997. Available from: http://rsb.info.nih.gov/ij/ |
| [20] | Snook JH, Li J, Helmke BP, et al. (2008) Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Radic Biol Med 44: 14-23. |
| [21] | Harris DE, Warshaw DM (1993) Smooth and skeletal muscle actin are mechanically indistinguishable in the in vitro motility assay. Circ Res 72: 219-224. |
| [22] | Bantscheff M, Schirle M, Sweetman G, et al. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem 389: 1017-1031. |
| [23] | dos Remedios CG, Moens PDJ (1995) Actin and the actomyosin interface: a review. Biochim Biophys Acta 1228: 99-124. |
| [24] | Drewes G, Faulstich H (1991) A reversible conformational transition in muscle actin is caused by nucleotide exchange and uncovers cysteine in position 10. J Biol Chem 266: 5508-5513. |
| [25] | Barden JA, Phillips L, Cornell BA, et al. (1989) Fluorine-19 NMR studies of the interaction of selectively labeled actin and myosin. Biochemistry 28: 5895-5901. |
| [26] | Lassing I, Schmitzberger F, Björnstedt M, et al. (2007) Molecular and Structural Basis for Redox Regulation of [beta]-Actin. J Mol Biol 370: 331-348. |
| [27] | Aldini G, Dalle-Donne I, Vistoli G, et al. (2005) Covalent modification of actin by 4-hydroxy-trans-2-nonenal (HNE): LC-ESI-MS/MS evidence for Cys374 Michael adduction. J Mass Spectrom 40: 946-954. |
| [28] | Johansson M, Lundberg M (2007) Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC Biochem 8: 26. |
| [29] | Dalle-Donne I, Giustarini D, Rossi R, et al. (2003) Reversible S-glutathionylation of Cys374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med 34: 23-32. |
| [30] | Vikhoreva NN, Vikhorev PG, Fedorova MA, et al. (2009) The in vitro motility assay parameters of actin filaments from Mytilus edulis exposed in vivo to copper ions. Arch Biochem Biophys 491: 32-38. |
| [31] | Wang J, Boja ES, Tan W, et al. (2001) Reversible Glutathionylation Regulates Actin Polymerization in A431 Cells. J Biol Chem 276: 47763-47766. |
| [32] | Otterbein LR, Graceffa P, Dominguez R (2001) The Crystal Structure of Uncomplexed Actin in the ADP State. Science 293: 708-711. |
| [33] | Doulias P-T, Greene JL, Greco TM, et al. (2010) Structural Profiling of Endogenous S-Nitrosocysteine Residues Reveals Unique Features That Accommodate Diverse Mechanisms for Protein S-Nitrosylation. Proc Natl Acad Sci U S A 107: 16958-16963 |
| [34] | Marino SM, Gladyshev VN (2010) Structural Analysis of Cysteine S-Nitrosylation: A Modified Acid-Based Motif and the Emerging Role of Trans-Nitrosylation. J Mol Biol 395: 844-859. |
| [35] | Miki M, dos Remedios CG (1988) Fluorescence quenching studies of fluorescein attached to Lys-61 or Cys-374 in actin: effects of polymerization, myosin subfragment-1 binding, and tropomyosin-troponin binding. J Biochem 104: 232-235. |
| [36] | Sun M, Rose MB, Ananthanarayanan SK, et al. (2008) Characterization of the pre-force-generation state in the actomyosin cross-bridge cycle. Proc Natl Acad Sci U S A 105: 8631-8636. |
| [37] | Galkin VE, Orlova A, Schröder GF, et al. (2010) Structural polymorphism in F-actin. Nat Struct Mol Biol 17: 1318-1323. |
| [38] | Jégou A, Romet-Lemonne G (2016) Single Filaments to Reveal the Multiple Flavors of Actin. Biophys J 110: 2138-2146. |
| [39] | Orlova A, Egelman EH (1993) A Conformational Change in the Actin Subunit Can Change the Flexibility of the Actin Filament. J Mol Biol 232: 334-341. |
| [40] | Orlova A, Egelman EH (1997) Cooperative rigor binding of myosin to actin is a function of F-actin structure. J Mol Biol 265: 469-474. |
| [41] | Tokuraku K, Uyeda TQ (2001) Phalloidin affects the myosin-dependent sliding velocities of actin filaments in a bound-divalent cation dependent manner. J Muscle Res Cell Motil 22: 371-378. |
| [42] | Vikhorev PG, Vikhoreva NN, Månsson A (2008) Bending flexibility of actin filaments during motor-induced sliding. Biophys J 95: 5809-5819. |
| [43] | Harris DE, Warshaw DM (1993) Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem 268: 14764-14768. |
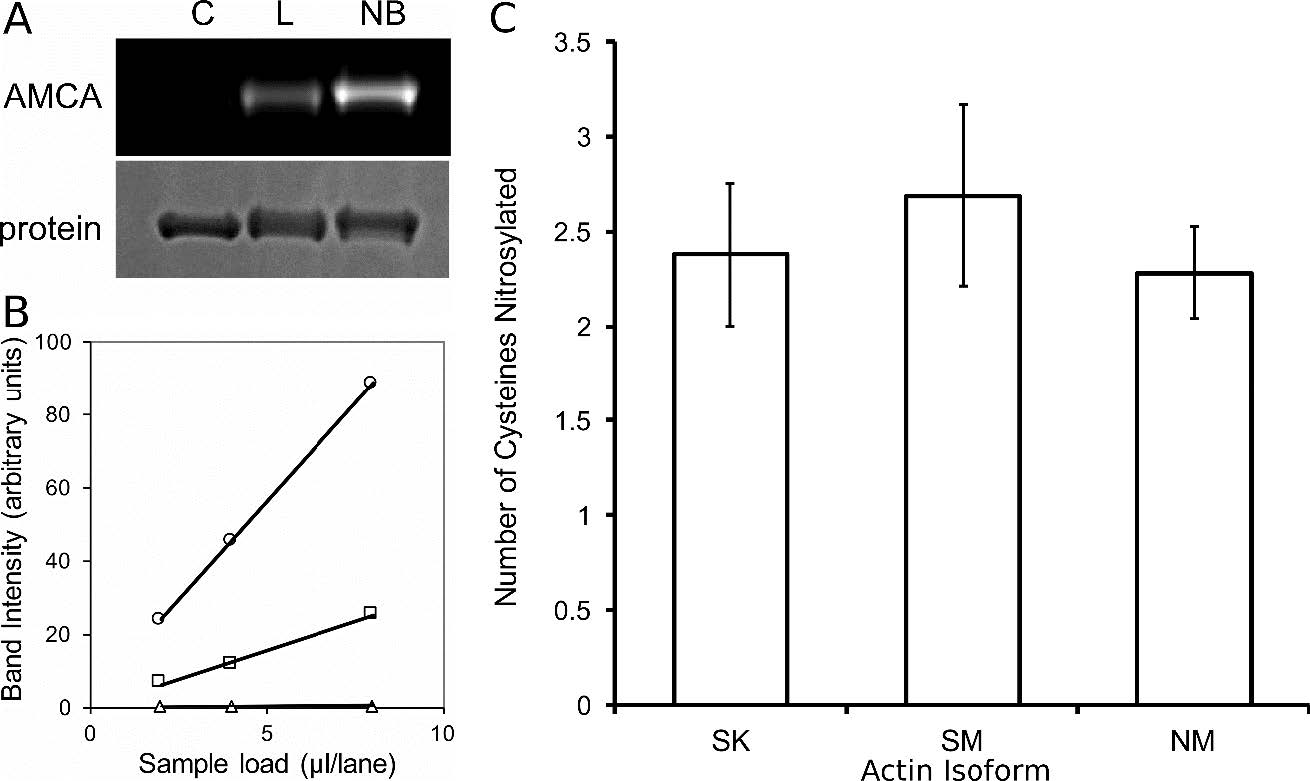
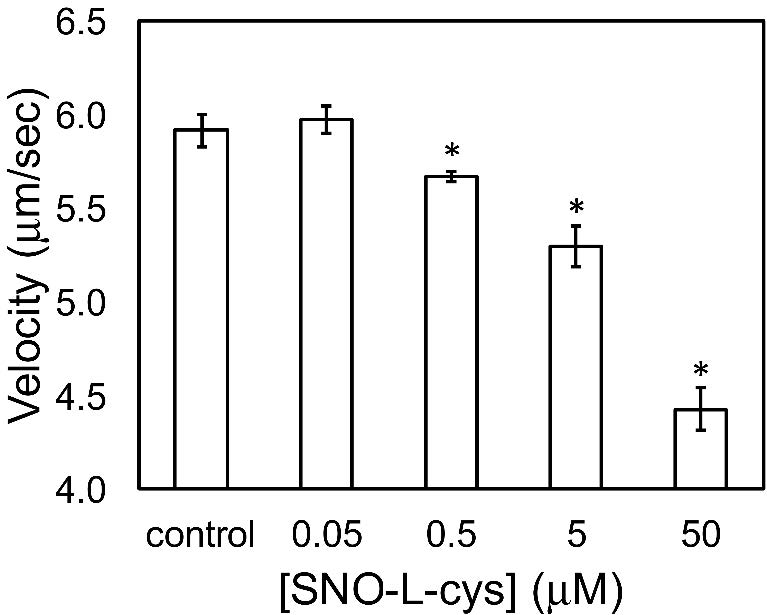
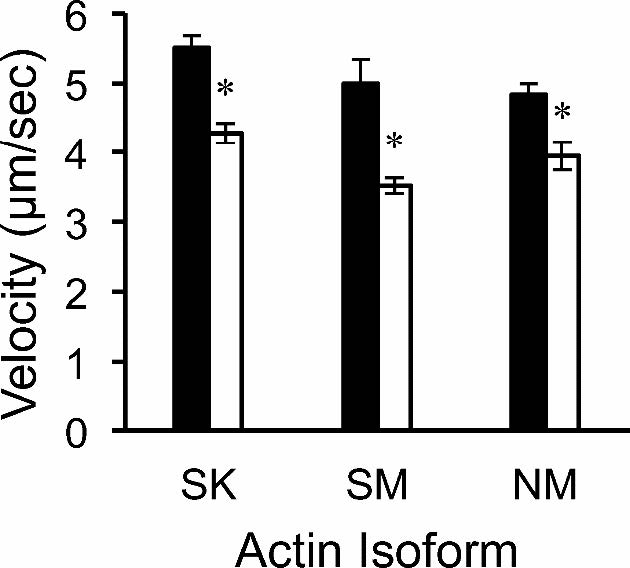
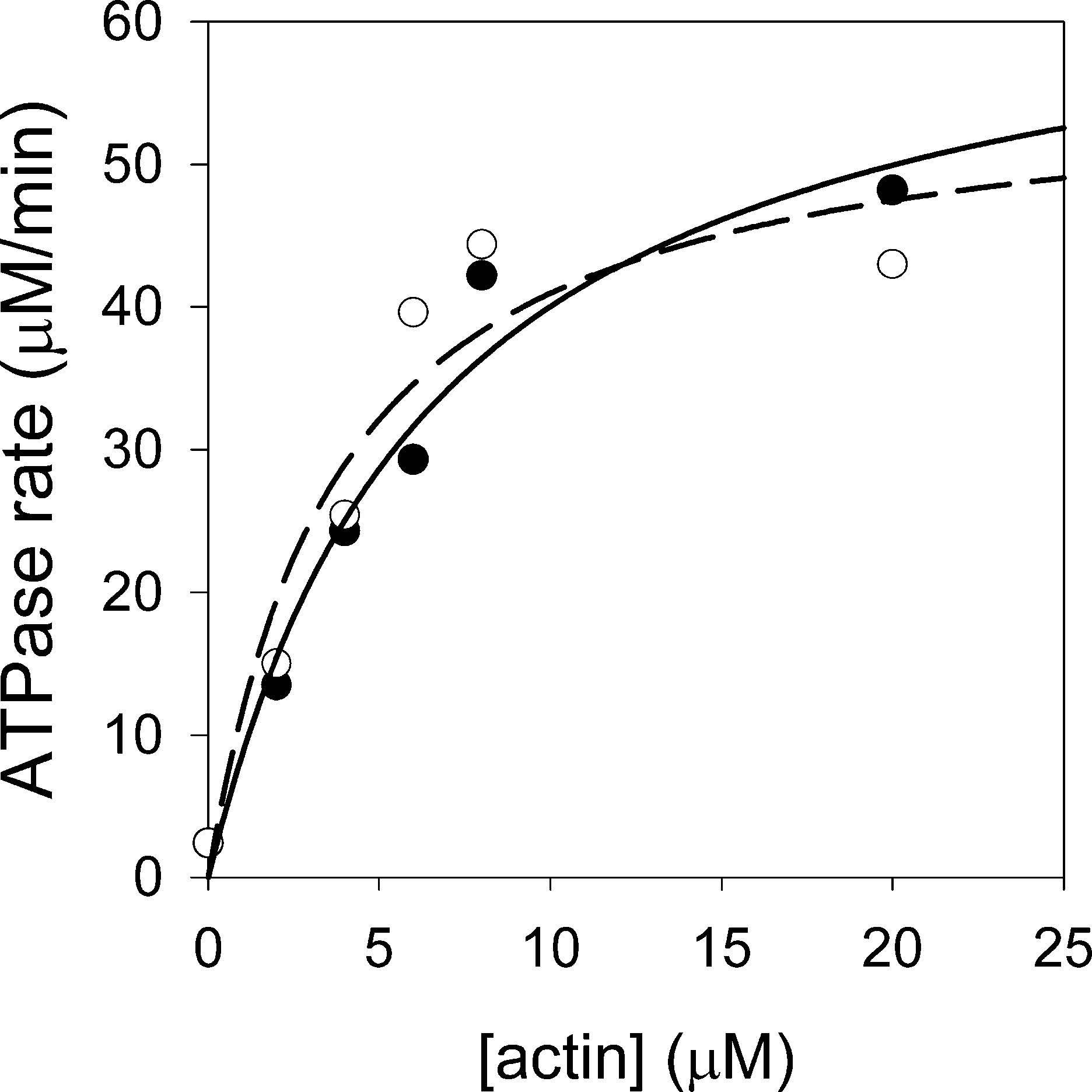
Figures(5) / Tables(1)

M.Bansbach Heather, H.Guilford William. Actin nitrosylation and its effect on myosin driven motility[J]. AIMS Molecular Science, 2016, 3(3): 426-438. doi: 10.3934/molsci.2016.3.426







 DownLoad:
DownLoad: