Citation: Achal Rastogi, Xin Lin, Bérangère Lombard, Damarys Loew, Leïla Tirichine. Probing the evolutionary history of epigenetic mechanisms: what can we learn from marine diatoms[J]. AIMS Genetics, 2015, 2(3): 173-191. doi: 10.3934/genet.2015.3.173

| [1] | Dolinoy DC (2008) The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev 66 Suppl 1: S7-11. |
| [2] |
Dolinoy DC (2007) Epigenetic gene regulation: early environmental exposures. Pharmacogenomics 8: 5-10. doi: 10.2217/14622416.8.1.5

|
| [3] |
Tariq M, Nussbaumer U, Chen Y, et al. (2009) Trithorax requires Hsp90 for maintenance of active chromatin at sites of gene expression. Proc Natl Acad Sci U S A 106: 1157-1162. doi: 10.1073/pnas.0809669106
|
| [4] |
Seong KH, Li D, Shimizu H, et al. (2011) Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145: 1049-1061. doi: 10.1016/j.cell.2011.05.029
|
| [5] |
Herrera CM, Bazaga P (2011) Untangling individual variation in natural populations: ecological, genetic and epigenetic correlates of long-term inequality in herbivory. Mol Ecol 20: 1675-1688. doi: 10.1111/j.1365-294X.2011.05026.x
|
| [6] | Dorrell RG, Smith AG (2011) Do red and green make brown?: perspectives on plastid acquisitions within chromalveolates. Eukaryot Cell 10: 856-868. |
| [7] |
Walker G, Dorrell RG, Schlacht A, et al. (2011) Eukaryotic systematics: a user's guide for cell biologists and parasitologists. Parasitology 138: 1638-1663. doi: 10.1017/S0031182010001708
|
| [8] |
Archibald JM (2009) The puzzle of plastid evolution. Curr Biol 19: R81-88. doi: 10.1016/j.cub.2008.11.067
|
| [9] |
Moustafa A, Beszteri B, Maier UG, et al. (2009) Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324: 1724-1726. doi: 10.1126/science.1172983
|
| [10] |
Bowler C, Allen AE, Badger JH, et al. (2008) The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456: 239-244. doi: 10.1038/nature07410
|
| [11] |
Amin SA, Parker MS, Armbrust EV (2012) Interactions between diatoms and bacteria. Microbiol Mol Biol Rev 76: 667-684. doi: 10.1128/MMBR.00007-12
|
| [12] | Falkowski PG, Barber RT, Smetacek VV (1998) Biogeochemical Controls and Feedbacks on Ocean Primary Production. Science 281: 200-207. |
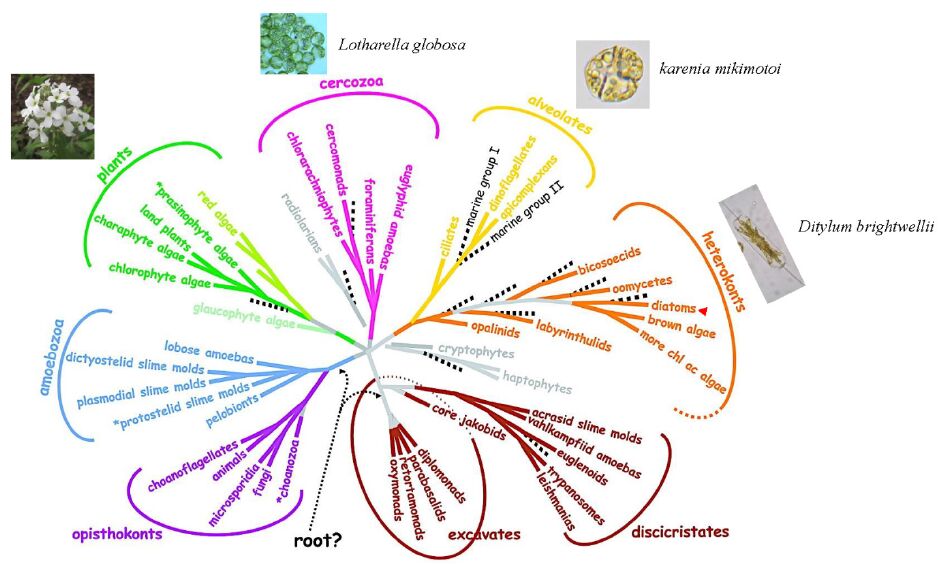
| [13] | Baldauf SL (2008) An overview of the phylogeny and diversity of eukaryotes. J Syst Evol 46: 263-273. |
| [14] |
Armbrust EV, Berges JA, Bowler C, et al. (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306: 79-86. doi: 10.1126/science.1101156
|
| [15] |
Lommer M, Specht M, Roy AS, et al. (2012) Genome and low-iron response of an oceanic diatom adapted to chronic iron limitation. Genome Biol 13: R66. doi: 10.1186/gb-2012-13-7-r66
|
| [16] |
Tanaka T, Maeda Y, Veluchamy A, et al. (2015) Oil Accumulation by the Oleaginous Diatom Fistulifera solaris as Revealed by the Genome and Transcriptome. Plant Cell 27: 162-176. doi: 10.1105/tpc.114.135194
|
| [17] |
Bowler C, De Martino A, Falciatore A (2010) Diatom cell division in an environmental context. Curr Opin Plant Biol 13: 623-630. doi: 10.1016/j.pbi.2010.09.014
|
| [18] |
Allen AE, Dupont CL, Obornik M, et al. (2011) Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature 473: 203-207. doi: 10.1038/nature10074
|
| [19] |
Tirichine L, Bowler C (2011) Decoding algal genomes: tracing back the history of photosynthetic life on Earth. Plant J 66: 45-57. doi: 10.1111/j.1365-313X.2011.04540.x
|
| [20] |
Maumus F, Allen AE, Mhiri C, et al. (2009) Potential impact of stress activated retrotransposons on genome evolution in a marine diatom. BMC Genomics 10: 624. doi: 10.1186/1471-2164-10-624
|
| [21] |
Lin X, Tirichine L, Bowler C (2012) Protocol: Chromatin immunoprecipitation (ChIP) methodology to investigate histone modifications in two model diatom species. Plant Methods 8: 48. doi: 10.1186/1746-4811-8-48
|
| [22] |
Tirichine L, Lin X, Thomas Y, et al. (2014) Histone extraction protocol from the two model diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Mar Genomics 13: 21-25. doi: 10.1016/j.margen.2013.11.006
|
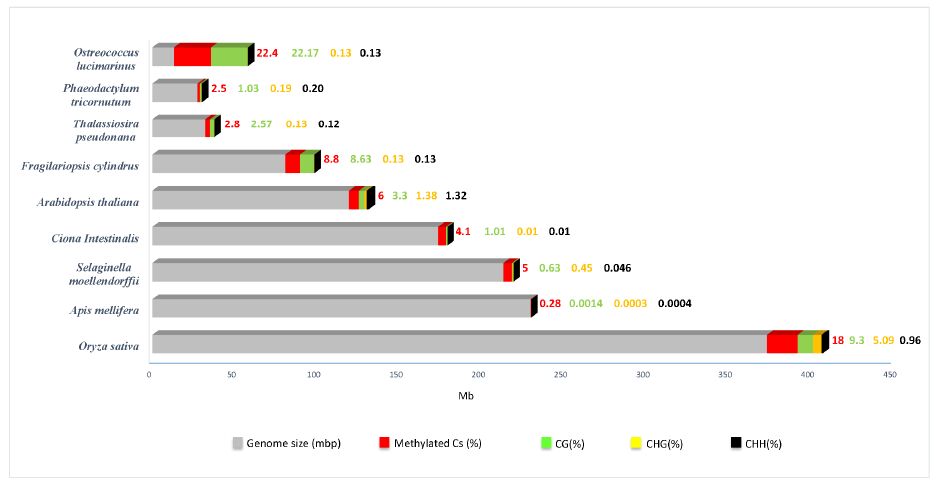
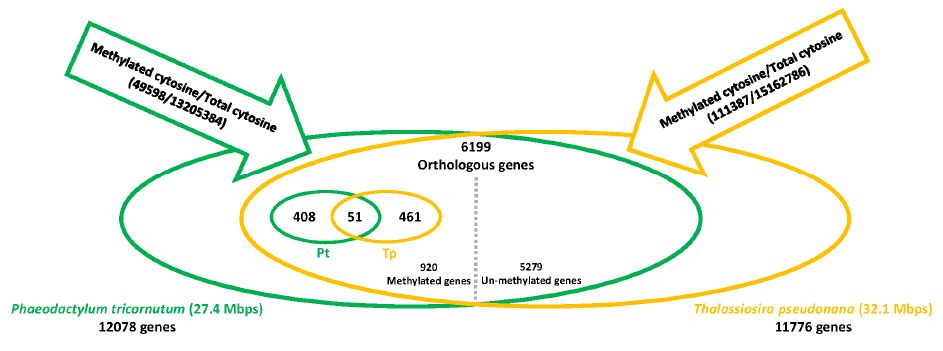
| [23] | Veluchamy A, Lin X, Maumus F, et al. (2013) Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat Commun 4: 2091. |
| [24] |
Veluchamy A, Rastogi A, Lin X, et al. (2015) An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol 16: 102. doi: 10.1186/s13059-015-0671-8
|
| [25] |
Rogato A, Richard H, Sarazin A, et al. (2014) The diversity of small non-coding RNAs in the diatom Phaeodactylum tricornutum. BMC Genomics 15: 698. doi: 10.1186/1471-2164-15-698
|
| [26] |
Huang A, He L, Wang G (2011) Identification and characterization of microRNAs from Phaeodactylum tricornutum by high-throughput sequencing and bioinformatics analysis. BMC Genomics 12: 337. doi: 10.1186/1471-2164-12-337
|
| [27] |
Cock JM, Sterck L, Rouze P, et al. (2010) The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465: 617-621. doi: 10.1038/nature09016
|
| [28] | Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465-476. |
| [29] |
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204-220. doi: 10.1038/nrg2719
|
| [30] |
Feng S, Cokus SJ, Zhang X, et al. (2010) Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 107: 8689-8694. doi: 10.1073/pnas.1002720107
|
| [31] |
Zemach A, McDaniel IE, Silva P, et al. (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916-919. doi: 10.1126/science.1186366
|
| [32] |
Goll MG, Bestor TH (2005) Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481-514. doi: 10.1146/annurev.biochem.74.010904.153721
|
| [33] |
Huff JT, Zilberman D (2014) Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 156: 1286-1297. doi: 10.1016/j.cell.2014.01.029
|
| [34] |
Zhang X, Yazaki J, Sundaresan A, et al. (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126: 1189-1201. doi: 10.1016/j.cell.2006.08.003
|
| [35] |
Molaro A, Hodges E, Fang F, et al. (2011) Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029-1041. doi: 10.1016/j.cell.2011.08.016
|
| [36] |
Zeng J, Konopka G, Hunt BG, et al. (2012) Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet 91: 455-465. doi: 10.1016/j.ajhg.2012.07.024
|
| [37] |
Honeybee Genome Sequencing C (2006) Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443: 931-949. doi: 10.1038/nature05260
|
| [38] |
Satou Y, Mineta K, Ogasawara M, et al. (2008) Improved genome assembly and evidence-based global gene model set for the chordate Ciona intestinalis: new insight into intron and operon populations. Genome Biol 9: R152. doi: 10.1186/gb-2008-9-10-r152
|
| [39] |
Goll MG, Kirpekar F, Maggert KA, et al. (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395-398. doi: 10.1126/science.1120976
|
| [40] |
Ponger L, Li WH (2005) Evolutionary diversification of DNA methyltransferases in eukaryotic genomes. Mol Biol Evol 22: 1119-1128. doi: 10.1093/molbev/msi098
|
| [41] |
Maumus F, Rabinowicz P, Bowler C, et al. (2011) Stemming Epigenetics in Marine Stramenopiles. Current Genomics 12: 357-370. doi: 10.2174/138920211796429727
|
| [42] |
Bowler C, Vardi A, Allen AE (2010) Oceanographic and biogeochemical insights from diatom genomes. Ann Rev Mar Sci 2: 333-365. doi: 10.1146/annurev-marine-120308-081051
|
| [43] | Zimmermann C, Guhl E, Graessmann A (1997) Mouse DNA methyltransferase (MTase) deletion mutants that retain the catalytic domain display neither de novo nor maintenance methylation activity in vivo. Biol Chem 378: 393-405. |
| [44] |
Fatemi M, Hermann A, Pradhan S, et al. (2001) The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J Mol Biol 309: 1189-1199. doi: 10.1006/jmbi.2001.4709
|
| [45] |
Penterman J, Zilberman D, Huh JH, et al. (2007) DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A 104: 6752-6757. doi: 10.1073/pnas.0701861104
|
| [46] |
Cokus SJ, Feng S, Zhang X, et al. (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215-219. doi: 10.1038/nature06745
|
| [47] | Hunt BG, Brisson JA, Yi SV, et al. Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol Evol 2: 719-728. |
| [48] |
Foret S, Kucharski R, Pittelkow Y, et al. (2009) Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes. BMC Genomics 10: 472. doi: 10.1186/1471-2164-10-472
|
| [49] |
Xiang H, Zhu J, Chen Q, et al. (2010) Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol 28: 516-520. doi: 10.1038/nbt.1626
|
| [50] |
Hellman A, Chess A (2007) Gene body-specific methylation on the active X chromosome. Science 315: 1141-1143. doi: 10.1126/science.1136352
|
| [51] | Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 6: e14524. |
| [52] |
Maunakea AK, Nagarajan RP, Bilenky M, et al. (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253-257. doi: 10.1038/nature09165
|
| [53] |
Takuno S, Gaut BS (2012) Body-Methylated Genes in Arabidopsis thaliana Are Functionally Important and Evolve Slowly. Mol Biol Evol 29: 219-227. doi: 10.1093/molbev/msr188
|
| [54] |
Zilberman D, Gehring M, Tran RK, et al. (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39: 61-69. doi: 10.1038/ng1929
|
| [55] |
Lorincz MC, Dickerson DR, Schmitt M, et al. (2004) Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 11: 1068-1075. doi: 10.1038/nsmb840
|
| [56] |
Luco RF, Pan Q, Tominaga K, et al. (2010) Regulation of alternative splicing by histone modifications. Science 327: 996-1000. doi: 10.1126/science.1184208
|
| [57] |
Lyko F, Foret S, Kucharski R, et al. (2010) The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol 8: e1000506. doi: 10.1371/journal.pbio.1000506
|
| [58] | Ammar R, Torti D, Tsui K, et al. (2012) Chromatin is an ancient innovation conserved between Archaea and Eukarya. Elife 1: e00078. |
| [59] |
Nalabothula N, Xi L, Bhattacharyya S, et al. (2013) Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genomics 14: 391. doi: 10.1186/1471-2164-14-391
|
| [60] |
Mersfelder EL, Parthun MR (2006) The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res 34: 2653-2662. doi: 10.1093/nar/gkl338
|
| [61] |
Cosgrove MS, Boeke JD, Wolberger C (2004) Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol 11: 1037-1043. doi: 10.1038/nsmb851
|
| [62] |
Lermontova I, Schubert V, Fuchs J, et al. (2006) Loading of Arabidopsis centromeric histone CENH3 occurs mainly during G2 and requires the presence of the histone fold domain. Plant Cell 18: 2443-2451. doi: 10.1105/tpc.106.043174
|
| [63] |
Hashimoto H, Sonoda E, Takami Y, et al. (2007) Histone H1 variant, H1R is involved in DNA damage response. DNA Repair (Amst) 6: 1584-1595. doi: 10.1016/j.dnarep.2007.05.003
|
| [64] |
Maheswari U, Jabbari K, Petit JL, et al. (2010) Digital expression profiling of novel diatom transcripts provides insight into their biological functions. Genome Biol 11: R85. doi: 10.1186/gb-2010-11-8-r85
|
| [65] |
Bheda P, Swatkoski S, Fiedler KL, et al. (2012) Biotinylation of lysine method identifies acetylated histone H3 lysine 79 in Saccharomyces cerevisiae as a substrate for Sir2. Proc Natl Acad Sci U S A 109: E916-925. doi: 10.1073/pnas.1121471109
|
| [66] |
Zhang K, Sridhar VV, Zhu J, et al. (2007) Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS One 2: e1210. doi: 10.1371/journal.pone.0001210
|
| [67] |
Tan M, Luo H, Lee S, et al. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146: 1016-1028. doi: 10.1016/j.cell.2011.08.008
|
| [68] |
Sana J, Faltejskova P, Svoboda M, et al. (2012) Novel classes of non-coding RNAs and cancer. J Transl Med 10: 103. doi: 10.1186/1479-5876-10-103
|
| [69] |
Ulitsky I, Bartel DP (2013) lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26-46. doi: 10.1016/j.cell.2013.06.020
|
| [70] |
Okazaki Y, Furuno M, Kasukawa T, et al. (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563-573. doi: 10.1038/nature01266
|
| [71] |
Cabili MN, Trapnell C, Goff L, et al. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915-1927. doi: 10.1101/gad.17446611
|
| [72] |
Liu J, Jung C, Xu J, et al. (2012) Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 24: 4333-4345. doi: 10.1105/tpc.112.102855
|
| [73] |
Pauli A, Valen E, Lin MF, et al. (2012) Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22: 577-591. doi: 10.1101/gr.133009.111
|
| [74] |
Cech TR, Steitz JA (2014) The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157: 77-94. doi: 10.1016/j.cell.2014.03.008
|
| [75] |
Molnar A, Schwach F, Studholme DJ, et al. (2007) miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447: 1126-1129. doi: 10.1038/nature05903
|
| [76] |
Zhao T, Li G, Mi S, et al. (2007) A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev 21: 1190-1203. doi: 10.1101/gad.1543507
|
| [77] |
Lopez-Gomollon S, Beckers M, Rathjen T, et al. (2014) Global discovery and characterization of small non-coding RNAs in marine microalgae. BMC Genomics 15: 697. doi: 10.1186/1471-2164-15-697
|
| [78] |
Norden-Krichmar TM, Allen AE, Gaasterland T, et al. (2011) Characterization of the small RNA transcriptome of the diatom, Thalassiosira pseudonana. PLoS One 6: e22870. doi: 10.1371/journal.pone.0022870
|
| [79] |
Supek F, Bosnjak M, Skunca N, et al. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800. doi: 10.1371/journal.pone.0021800
|
Figures(7) / Tables(2)

Achal Rastogi, Xin Lin, Bérangère Lombard, Damarys Loew, Leïla Tirichine. Probing the evolutionary history of epigenetic mechanisms: what can we learn from marine diatoms[J]. AIMS Genetics, 2015, 2(3): 173-191. doi: 10.3934/genet.2015.3.173







 DownLoad:
DownLoad: