Citation: Sergio Branciamore, Andrei S. Rodin, Grigoriy Gogoshin, Arthur D. Riggs. Epigenetics and Evolution: Transposons and the Stochastic Epigenetic Modification Model[J]. AIMS Genetics, 2015, 2(2): 148-162. doi: 10.3934/genet.2015.2.148

| [1] | M. B. Ekram, K. Kang, H. Kim and J. Kim Retrotransposons as a major source of epigenetic variations in the mammalian genome. Epigenetics : official journal of the DNA Methylation Society, 7 (2012), 370-382. |
| [2] | T. H. Bestor, J. R. Edwards and M. Boulard Notes on the role of dynamic DNA methylation in mammalian development. Proceedings of the National Academy of Sciences of the United States of America,(2014), in press. |
| [3] | M. Thomas, L. Pingault, A. Poulet, J. Duarte, M. Throude, S. Faure, J. P. Pichon, E. Paux, A. V. Probst and C. Tatout Evolutionary history of Methyltransferase 1 genes in hexaploid wheat. BMC genomics, 15 (2014), 922. |
| [4] | H. L. Levin and J. V. Moran Dynamic interactions between transposable elements and their hosts. Nature reviews. Genetics,12 (2011), 615-627. |
| [5] | C. Feschotte Transposable elements and the evolution of regulatory networks. Nature reviews. Genetics,9 (2008),397-39405. |
| [6] | E. Heard and R. A. Martienssen Transgenerational epigenetic inheritance: myths and mechanisms. Cell,157 (2014),95-95109. |
| [7] | C. F. Kratochwil and A. Meyer Closing the genotype-phenotype gap: emerging technologies for evolutionary genetics in ecological model vertebrate systems. BioEssays : news and reviews in molecular, cellular and developmental biology,37 (2015), 213-226. |
| [8] | A. P. Feinberg and R. A. Irizarry Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proceedings of the National Academy of Sciences of the United States of America, 107 Suppl 1 (2010), 1757-64. |
| [9] | E. Jablonka Epigenetic variations in heredity and evolution. Clinical pharmacology and therapeutics, 92 (2012),683-8. |
| [10] | T. E. Keller and S. V. Yi DNA methylation and evolution of duplicate genes. Proceedings of the National Academy of Sciences of the United States of America, 111 (2014), 5932-7. |
| [11] | M. M. Darby and S. Sabunciyan Repetitive elements and epigenetic marks in behavior and psychiatric disease. Advances in genetics, 86 (2014), 185-252. |
| [12] | S. Branciamore, A. S. Rodin, A. D. Riggs and S. N. Rodin Enhanced evolution by stochastically variable modification of epigenetic marks in the early embryo. Proceedings of the National Academy of Sciences of the United States of America, 111 (2014), 6353-8. |
| [13] | S. N. Rodin and A. D. Riggs Epigenetic silencing may aid evolution by gene duplication. Journal of molecular evolution, 56 (2003), 718-29. |
| [14] | S. N. Rodin, D. V. Parkhomchuk, A. S. Rodin, G. P. Holmquist and A. D. Riggs Repositioning-dependent fate of duplicate genes. DNA and cell biology, 24 (2005), 529-42. |
| [15] | M. R. Reed, A. D. Riggs and J. R. Mann Deletion of a direct repeat element has no effect on Igf2 and H19 imprinting. Mammalian genome, 12 (2001), 873-6. |
| [16] | A. Chess Mechanisms and consequences of widespread random monoallelic expression. Nature reviews. Genetics, 13 (2012), 421-8. |
| [17] | A. M. Cotton, E. M. Price, M. J. Jones, B. P. Balaton, M. S. Kobor and C. J. Brown Landscape of DNA methylation on the X chromosome reffects CpG density, functional chromatin state and X-chromosome inactivation. Human molecular genetics,(2014), in press. |
| [18] | A. V. Gendrel, M. Attia, C. J. Chen, P. Diabangouaya, N. Servant, E. Barillot and E. Heard Developmental dynamics and disease potential of random monoallelic gene expression. Developmental cell, 28 (2014), 366-80. |
| [19] | A. A. Adegbola, G. F. Cox, E. M. Bradshaw, D. A. Ha er, A. Gimelbrant and A. Chess Monoallelic expression of the human FOXP2 speech gene. Proceedings of the National Academy of Sciences of the United States of America,(2014), in press. |
| [20] | M. A. Eckersley-Maslin, D. Thybert, J. H. Bergmann, J. C. Marioni, P. Flicek and D. L. Spector Random monoallelic gene expression increases upon embryonic stem cell differentiation. Developmental cell, 28 (2014), 351-65. |
| [21] | J. P. Shonkoff et. al. Early Experiences Can Alter Gene Expression and Affect Long-Term Development: Working Paper No. 10. National Scientific Council on the Developing Child (2010). |
| [22] | T. Tollefsbol Handbook of Epigenetics, The New Molecular and Medical Genetics 1st edition, Academic Press San Diego, 2011 |
| [23] | C. Weinhouse, O. S. Anderson, T. R. Jones, J. Kim, S. A. Liberman, M. S. Nahar, L. S. Rozek, R. L. Jirtle and D. C. Dolinoy An expression microarray approach for the identification of metastable epialleles in the mouse genome. Epigenetics : official journal of the DNA Methylation Society, 6 (2011), 1105-13. |
| [24] | D. C. Dolinoy, C. Weinhouse, T. R. Jones, L. S. Rozek and R. L. Jirtle Variable histone modifications at the A(vy) metastable epiallele. Epigenetics, 5 (2010), 637-44. |
| [25] | M. Francescatto, M. Vitezic, P. Heutink and A. Saxena Brain-specific noncoding RNAs are likely to originate in repeats and may play a role in up-regulating genes in cis. The international journal of biochemistry & cell biology,54 (2014), 331-7. |
| [26] | E. Maclary, E. Buttigieg, M. Hinten, S. Gayen, C. Harris, M. K. Sarkar, S. Purushothaman and S. Kalantry Differentiation-dependent requirement of Tsix long non-coding RNA in imprinted X-chromosome inactivation. Nature communications, 5 (2014), 4209. |
| [27] | J. H. Thomas and S. Schneider Coevolution of retroelements and tandem zinc finger genes. Genome research, 21 (2011), 1800-12. |
| [28] | Z. D. Smith, M. M. Chan, T. S. Mikkelsen, H. Gu, A. Gnirke, A. Regev and A. Meissner A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature, 484 (2012), 339-44. |
| [29] | H. Guo, P. Zhu, L. Yan, R. Li, B. Hu, Y. Lian, J. Yan, X. Ren, S. Lin, J. Li, X. Jin, X. Shi, P. Liu, X. Wang, W. Wang, Y. Wei, X. Li, F. Guo, X. Wu, X. Fan, J. Yong, L. Wen, S. X. Xie, F. Tang and J. Qiao The DNA methylation landscape of human early embryos. Nature, 511 (2014), 606-10. |
| [30] | Z. D. Smith, M. M. Chan, K. C. Humm, R. Karnik, S. Mekhoubad, A. Regev, K. Eggan and A. Meissner DNA methylation dynamics of the human preimplantation embryo. Nature, 511 (2014), 611-5. |
| [31] | Z. X. Chen and A. D. Riggs DNA methylation and demethylation in mammals. The Journal of biological chemistry,286 (2011), 18347-53. |
| [32] | S. B. Baylin and P. A. Jones A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews. Cancer, 11 (2011), 726-34. |
| [33] | Z. D. Smith and A. Meissner DNA methylation: roles in mammalian development. Nature reviews. Genetics,14 (2013), 204-220. |
| [34] | S. M. Cullen, A. Mayle, L. Rossi and M. A. Goodell Hematopoietic stem cell development: an epigenetic journey. Current topics in developmental biology, 107 (2014), 39-75. |
| [35] | M. Harris High-frequency induction by 5-azacytidine of proline independence in CHO-K1 cells. Somatic cell and molecular genetics, 10 (1984), 615-24. |
| [36] | Y. Shen, F. Yue, D. F. McCleary, Z. Ye, L. Edsall, S. Kuan, U. Wagner, J. Dixon, L. Lee, V. V. Lobanenkov and B. Ren A map of the cis-regulatory sequences in the mouse genome. Nature, 488 (2012), 116-20. |
| [37] | S. C. Elgin and G. Reuter Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harbor perspectives in biology, 5 (2013), a017780. |
| [38] | L. H. Chadwick, L. M. Pertz, K. W. Broman, M. S. Bartolomei and H. F. Willard Genetic control of X chromosome inactivation in mice: definition of the Xce candidate interval. Genetics, 173 (2006), 2103-10. |
| [39] | A. L. Jones and S. Sung Mechanisms underlying epigenetic regulation in Arabidopsis thaliana. Integrative and comparative biology, 54 (2014), 61-7. |
| [40] | E. J. Kim, X. Ma and H. Cerutti Gene silencing in microalgae: Mechanisms and biological roles. Bioresource technology, (2014), in press. |
| [41] | P. T. West, Q. Li, L. Ji, S. R. Eichten, J. Song, M. W. Vaughn, R. J. Schmitz and N. M. Springer Genomic distribution of H3K9me2 and DNA methylation in a maize genome. PloS One, 9 (2014), e105267. |
| [42] | M. Y. Kim and D. Zilberman DNA methylation as a system of plant genomic immunity. Trends in plant science, 19 (2014), 320-6. |
| [43] | D. T. Ge and P. D. Zamore Small RNA-directed silencing: the y finds its inner fission yeast? Current biology, 23 (2013), R318-20. |
| [44] | Z. Guo, W. Jiang, N. Lages, W. Borcherds and D. Wang Relationship between gene duplicability and diversifiability in the topology of biochemical networks. BMC genomics, 15 (2014), 577. |
| [45] | A. N. Nguyen Ba, B. Strome, J. J. Hua, J. Desmond, I. Gagnon-Arsenault, E. L. Weiss, C. R. Landry and A. M. Moses Detecting Functional Divergence after Gene Duplication through Evolutionary Changes in Posttranslational Regulatory Sequences. PLoS computational biology, 10 (2014), e1003977. |
| [46] | A. S. Rodin, G. Gogoshin, A. Litvinenko, E. Boerwinkle Exploring Genetic Epidemiology Data with Bayesian Networks Handbook of Statistics, 28 (2012), 479-510 |
| [47] | Y. Zhang, J. Zhang and J. Shang Quantitative identification of differentially methylated loci based on relative entropy for matched case-control data. Epigenomics, 5 (2013), 631-43. |
| [48] | C. Faulk, A. Barks, K. Liu, J. M. Goodrich and D. C. Dolinoy Early-life lead exposure results in dose- and sex-specific effects on weight and epigenetic gene regulation in weanling miceu. Epigenomics, 5 (2013), 487-500. |
| [49] | J. A. Capra and D. Kostka Modeling DNA methylation dynamics with approaches from phylogenetics. Bioinformatics (Oxford, England), 30 (2014), i408-14. |
| [50] | A. Y. Chang and B. Y. Liao DNA methylation rebalances gene dosage after mammalian gene duplications. Molecular biology and evolution, 29 (2012), 133-44. |
| [51] | S. Toyoda, M. Kawaguchi, T. Kobayashi, E. Tarusawa, T. Toyama, M. Okano, M. Oda, H. Nakauchi, Y. Yoshimura, M. Sanbo, M. Hirabayashi, T. Hirayama, T. Hirabayashi and T. Yagi Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron, 82 (2014), 94-108. |
| [52] | C. Lorthongpanich, L. F. Cheow, S. Balu, S. R. Quake, B. B. Knowles, W. F. Burkholder, D. Solter and D. M. Messerschmidt Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science (New York, N.Y.), 341 (2013), 1110-2. |
| [53] | H. Guo, P. Zhu, X. Wu, X. Li, L. Wen and F. Tang Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome research, 23 (2013), 2126-35. |
| [54] | Z. S. Singer, J. Yong, J. Tischler, J. A. Hackett, A. Altinok, M. A. Surani, L. Cai and M. B. Elowitz Dynamic heterogeneity and DNA methylation in embryonic stem cells. Molecular cell, 55 (2014), 319-31. |
| [55] | J. A. Doudna and E. Charpentier Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science (New York, N.Y.), 346 (2014), 1258096. |
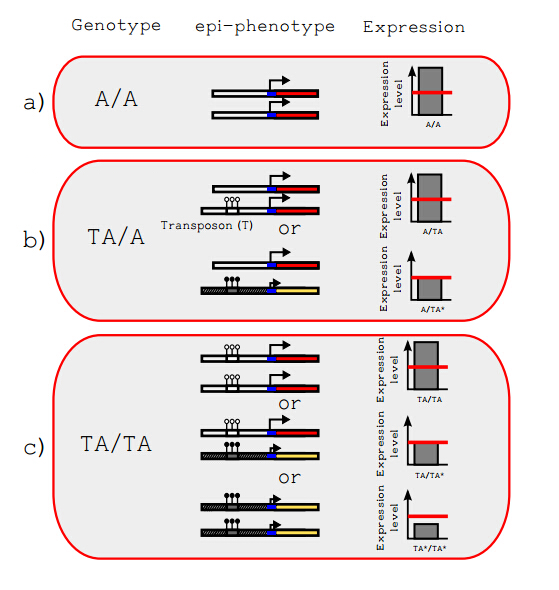
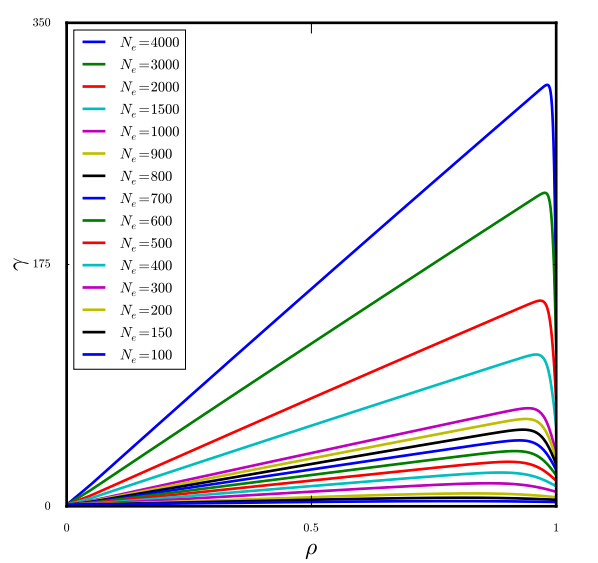
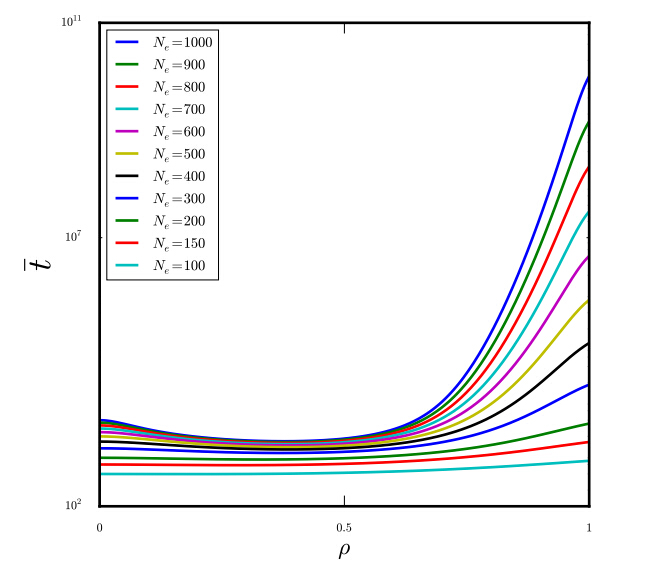
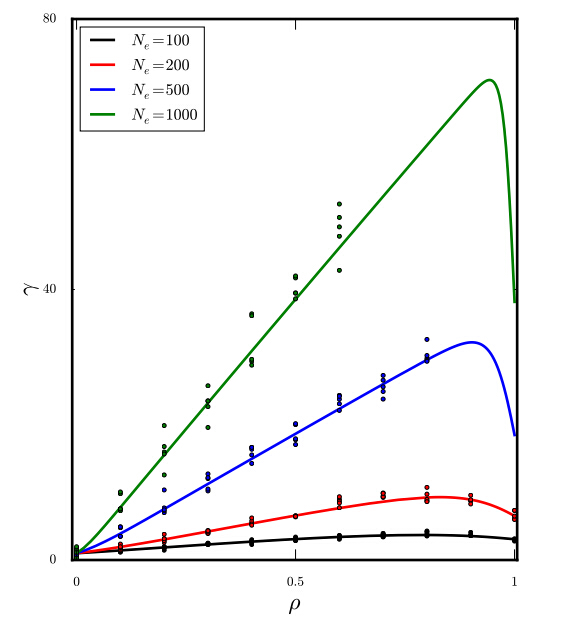
Figures(5)

Sergio Branciamore, Andrei S. Rodin, Grigoriy Gogoshin, Arthur D. Riggs. Epigenetics and Evolution: Transposons and the Stochastic Epigenetic Modification Model[J]. AIMS Genetics, 2015, 2(2): 148-162. doi: 10.3934/genet.2015.2.148







 DownLoad:
DownLoad: