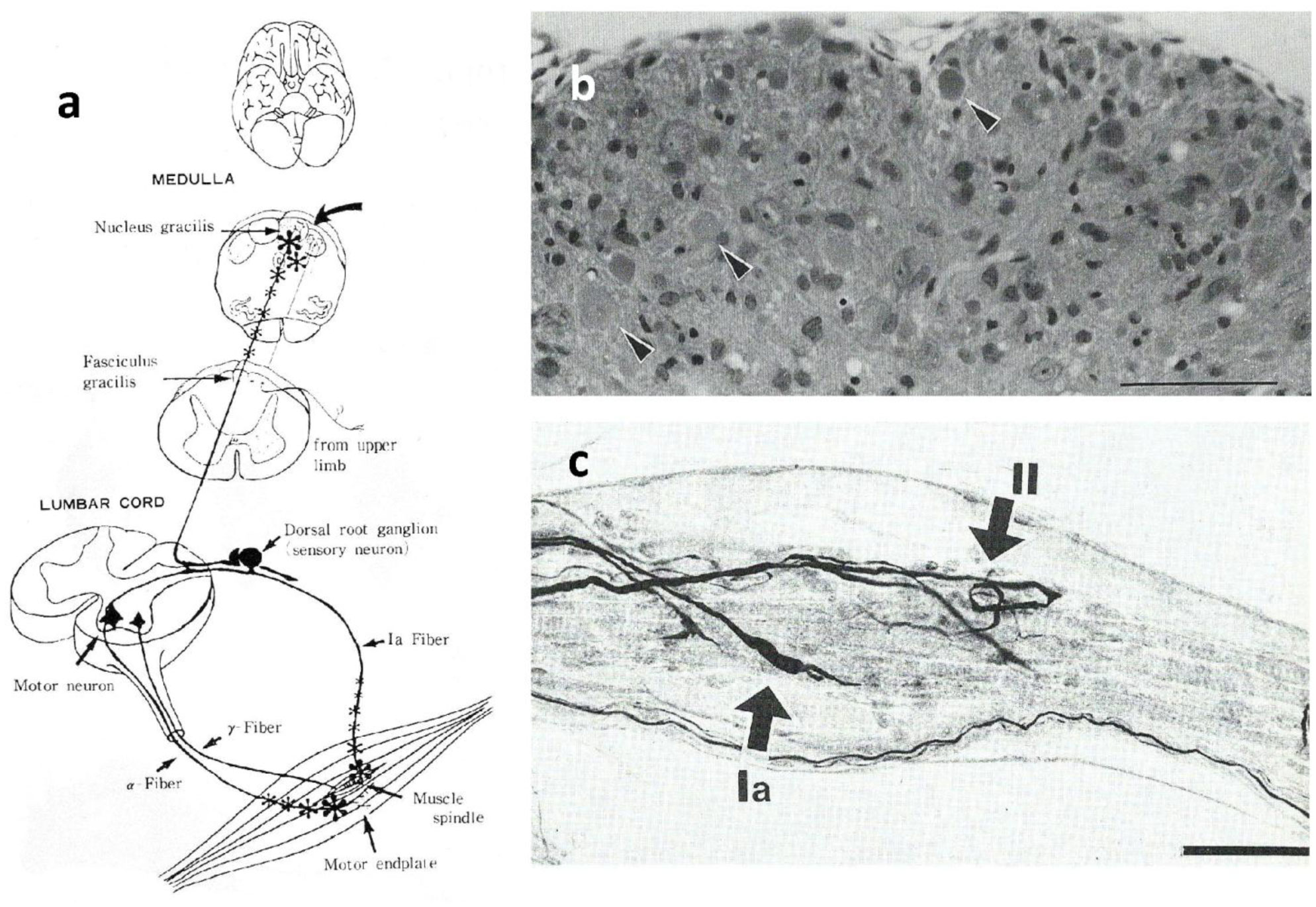
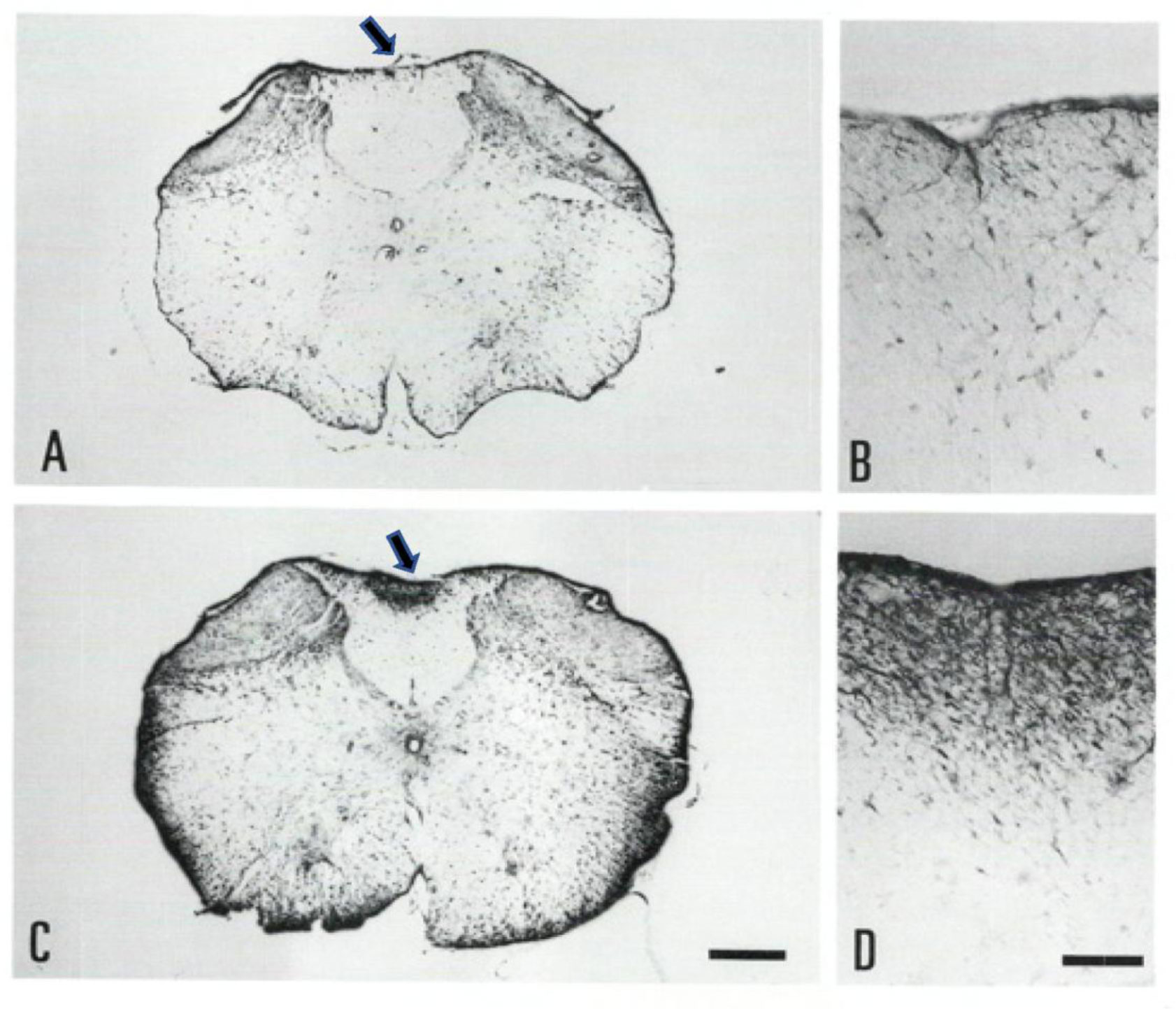
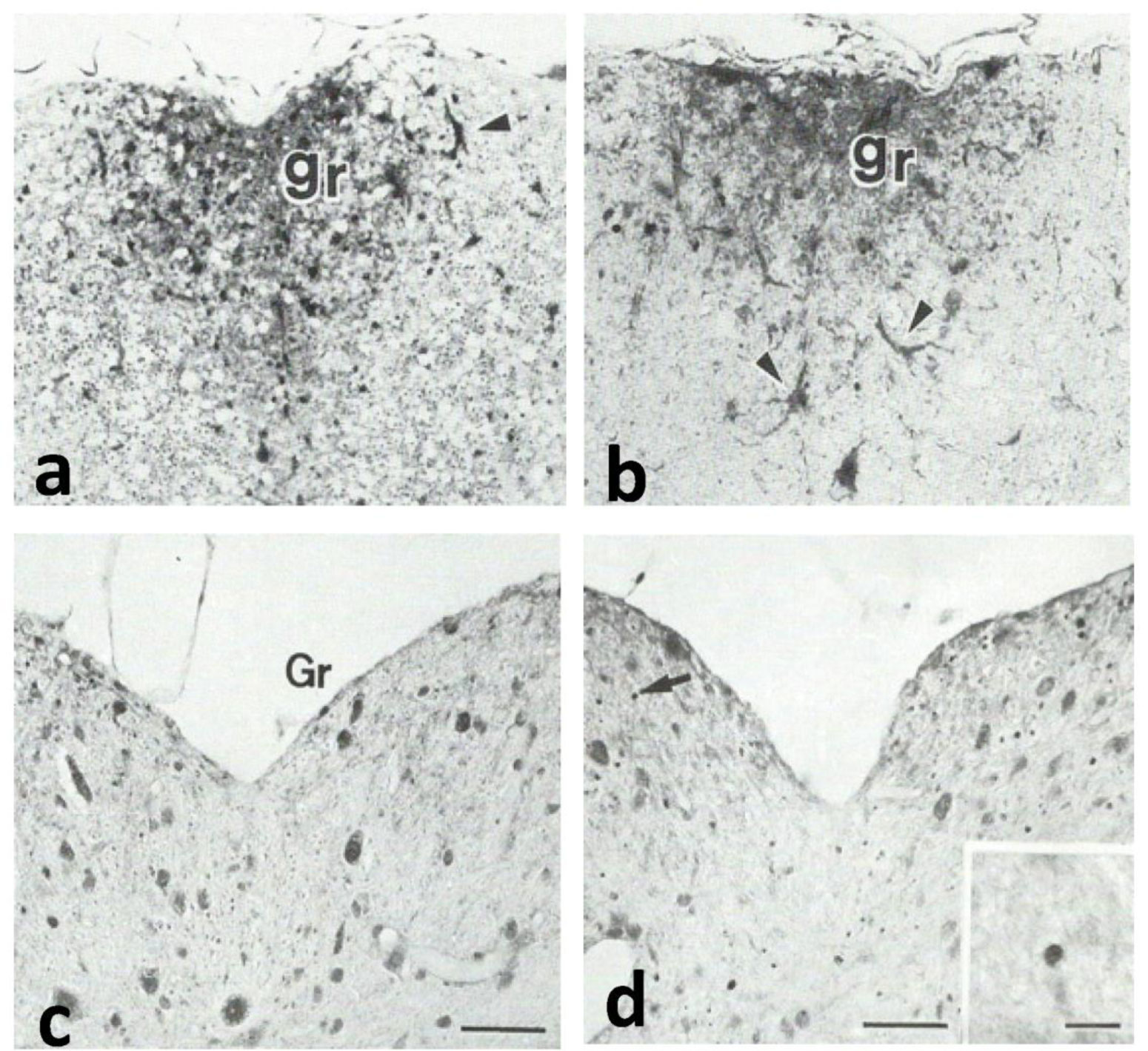
Gracile axonal dystrophy (gad) mouse shows tremor, ataxia and muscular atrophy of hind limbs from about 80-days of age. These clinical features become progressively severe to death. Pathological examination reveals that main and early axonal degeneration exists in a long ascending nervous tract in dorsal column of the spinal cord: gracile nucleus and fascicules. Similar lesions are seen in axonal terminals of peripheral sensory (muscle spindles) and motor endplates. Most striking features of axonal dystrophy are “dying-back” axonal degeneration with partial swellings (“spheroids” in matured type) which come to be most frequently in gracile nucleus, followed by in order of gracile fasciculus of cervical, thoracic and lumber cord levels. Immunocytochemical increase of glial fibrillary acidic protein (GFAP) and substance P (SP) is seen in reactive astrocytes and degenerating axons. Likewise, amyloid precursor protein (APP) and amyloid β-protein (AβP) activity become positive in axons and astrocytes along ascending tract. Moreover, ubiquitin-positive dot-like structures accumulate in gracile nucleus, spinocerebellar tract, and cerebellum in gad mice after 9th-week old. Ubiquitinated structures are localized in spheroids with a larger diameter than normal. The gad mutation is caused by an in-frame deletion including exon 7 and 8 of UCH-L1 gene, encoding the ubiquitin c-terminal hydrolase (UCH) isozyme (UCH-L1) selectively expressed in nervous system and testis/ovary. The gad allele encodes a truncated UCH-L1 lacking a segment of 42 amino acids containing catalytic site. The evaluation as mouse models for Parkinson's and Alzheimer's diseases and the collapse of synapse-axon circulation around central nervous system from peripheral nervous system are discussed.
Citation: Tateki Kikuchi. Circular breakdown of neural networks due to loss of deubiquitinating enzyme (UCH-L1) in gracile axonal dystrophy (gad) mouse[J]. AIMS Molecular Science, 2021, 8(4): 311-324. doi: 10.3934/molsci.2021024
Gracile axonal dystrophy (gad) mouse shows tremor, ataxia and muscular atrophy of hind limbs from about 80-days of age. These clinical features become progressively severe to death. Pathological examination reveals that main and early axonal degeneration exists in a long ascending nervous tract in dorsal column of the spinal cord: gracile nucleus and fascicules. Similar lesions are seen in axonal terminals of peripheral sensory (muscle spindles) and motor endplates. Most striking features of axonal dystrophy are “dying-back” axonal degeneration with partial swellings (“spheroids” in matured type) which come to be most frequently in gracile nucleus, followed by in order of gracile fasciculus of cervical, thoracic and lumber cord levels. Immunocytochemical increase of glial fibrillary acidic protein (GFAP) and substance P (SP) is seen in reactive astrocytes and degenerating axons. Likewise, amyloid precursor protein (APP) and amyloid β-protein (AβP) activity become positive in axons and astrocytes along ascending tract. Moreover, ubiquitin-positive dot-like structures accumulate in gracile nucleus, spinocerebellar tract, and cerebellum in gad mice after 9th-week old. Ubiquitinated structures are localized in spheroids with a larger diameter than normal. The gad mutation is caused by an in-frame deletion including exon 7 and 8 of UCH-L1 gene, encoding the ubiquitin c-terminal hydrolase (UCH) isozyme (UCH-L1) selectively expressed in nervous system and testis/ovary. The gad allele encodes a truncated UCH-L1 lacking a segment of 42 amino acids containing catalytic site. The evaluation as mouse models for Parkinson's and Alzheimer's diseases and the collapse of synapse-axon circulation around central nervous system from peripheral nervous system are discussed.
Alzheimer's disease
amyloid precursor protein
APP-immunoreactivity
adenosine triphosphate
amyloid β protein
AβP-immunoreactivity
cornu ammonis 1
cholecystokinin
central nervous system
dorsal root ganglia
gracile axonal dystrophy, mouse name by pathological features
gracile axonal dystrophy, mouse name after responsible gene was identified
glial fibrillary acidic protein
long-term potentiation
national center for neurology and psychiatry
Parkinson's disease
peripheral nervous system
substance P
ubiquitin
ubiquitin c-terminal hydrolase, isozyme L1

| [1] | Oda K, Yamazaki K, Miura H, et al. (1991) Gracile axonal dystrophy (GAD) mouse- An animal model of “dying back” type axonal degeneration-. Shinkei Kenkyu no Shinpo 35: 95-105. |
| [2] |
Yamazaki K, Wakasugi N, Tomita T, et al. (1988) Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proc Soc Exp Biol Med 187: 209-215. doi: 10.3181/00379727-187-42656

|
| [3] |
Yamazaki K, Sakakibara A, Tomita T, et al. (1987) Location of gracile axonal dystrophy (gad) on chromosome 5 of the mouse. Jpn J Genet 62: 479-484. doi: 10.1266/jjg.62.479
|
| [4] |
Kikuchi T, Mukoyama M, Yamazaki K, et al. (1990) Axonal degeneration of ascending sensory neurons in gracile axonal dystrophy mutant mouse. Acta Neuropathol 80: 145-151. doi: 10.1007/BF00308917
|
| [5] |
Oda K, Yamazaki K, Miura H, et al. (1992) Dying back type axonal degeneration of sensory nerve terminals in muscle spindles of the gracile axonal dystrophy (GAD) mutant mouse. Neuropathol Appl Neurobiol 18: 265-281. doi: 10.1111/j.1365-2990.1992.tb00789.x
|
| [6] | Fujisawa K (1967) A unique type of axonal alteration (so-called axonal dystrophy) as seen in Goll's nucleus of 277 cases of control: A contribution to the pathology of aging process. Acta Neurobiol 8: 255-275. |
| [7] |
Fujisawa K, Shiraki H (1978) Study of axonal dystrophy. I. Pathology of the neuronal of the gracile and the cuneate nuclei on ageing and old rats: a stereological study. Neuropath. Appl Neurobiol 4: 1-20. doi: 10.1111/j.1365-2990.1978.tb00525.x
|
| [8] |
Mukoyama M, Yamazaki K, Kikuchi T, et al. (1989) Neuropathology of gracile axonal dystrophy (GAD) mouse: An animal model of central distal axonopathy in primary sensoneurons. Acta Neuropathol 79: 294-299. doi: 10.1007/BF00294664
|
| [9] |
Kowalski JR, Juo P (2012) The role of deubiquitinating enzymes in synaptic function and nervous system diseases. Neural Plast 2012: 1-13. doi: 10.1155/2012/892749
|
| [10] |
Svoboda K, Christoph F, Schmidt BJ, et al. (1993) Direct observation of kinesin stepping by optical trapping interferometry. Nature 365: 721-727. doi: 10.1038/365721a0
|
| [11] |
Matsushima Y, Kikuchi T, Kikuchi H, et al. (2005) A new mouse model for infantile neuroaxonal dystrophy, inad mouse, maps to mouse Chromosome 1. Mamm Genome 16: 73-78. doi: 10.1007/s00335-004-3017-5
|
| [12] |
Sacks OW, Agular MJ, Brown WJ (1966) Hallervorden-Spatz disease: Its pathogenesis and place among the axonal dystrophies. Acta Neuripathol 6: 164-174. doi: 10.1007/BF00686761
|
| [13] |
Pentschew A, Schwarz K (1962) Systemic axonal dystrophy in vitamin E deficient adult rats: With implication in human neuropathology. Acta Neuropathol 1: 313-334. doi: 10.1007/BF00687729
|
| [14] |
Yamazaki KY, Mukoyama M, Kikuchi T, et al. (1989) Effects of dietary vitamin E supplement in Gracile Axonal Dystrophy (gad) mice. Exp Anim 38: 195-200. doi: 10.1538/expanim1978.38.3_195
|
| [15] |
Brannon W, McCormick W, Lampart P (1967) Axonal dystrophy in the gracile nucleus of man. Acta Neuropathol 9: 1-6. doi: 10.1007/BF00688153
|
| [16] |
Fujisawa K, Baek SY, Arakawa T, et al. (1995) Calcitonin gene-related peptide- and substance P-immunoreactive axons in the nucleus gracilis of the rat with special reference to axonal dystrophy: light and electron microscopic observations. Acta Neuropathol 90: 347-355. doi: 10.1007/BF00315008
|
| [17] |
Yoshikawa H, Tarui S, Hashimoto PH (1985) Diminished retrograde transport causes axonal dystrophy in the nucleus gracilis. Electron- and light-microscopic study. Acta Neuropathol 68: 93-100. doi: 10.1007/BF00688629
|
| [18] |
Miura H, Oda K, Endo C, et al. (1993) Progressive degeneration of motor nerve terminals in GAD mutant mouse with hereditary sensory axonopathy. Neuropathol Appl Neurobiol 19: 41-51. doi: 10.1111/j.1365-2990.1993.tb00403.x
|
| [19] |
Reinicke AT, Laban K, Schs M, et al. (2019) Ubiquitin c-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover on the first postnatal weeks. PNAS 116: 7963-7972. doi: 10.1073/pnas.1812413116
|
| [20] |
Mi W, Beirowski B, Gillingwarter TA, et al. (2005) The slow Wallerian degeneration gene, Wlds, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain 128: 405-416. doi: 10.1093/brain/awh368
|
| [21] | Takagi A, Kajiwara H, Oda K, et al. (1996) Fine structural changes of muscle spindles in the gracile axonal dystrophy mutant mouse. Virchows Achiv 428: 289-296. |
| [22] |
Adalbert R, Nogradi A, Babetto E, et al. (2009) Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132: 402-413. doi: 10.1093/brain/awn312
|
| [23] |
Genc B, Jara JH, Schults MC, et al. (2016) Absence of UCHL 1 function leads to selective motor neuropathy. Ann Clinic Translat Neurol 3: 331-345. doi: 10.1002/acn3.298
|
| [24] |
Coleman MR, Freeman MR (2010) Wallerian degeneration, Wlds, and Nmnat. Annual Rev Neurosci 33: 245-267. doi: 10.1146/annurev-neuro-060909-153248
|
| [25] |
Yamazaki K, Moriya H, Wakabayashi T, et al. (1989) Substance P-like immunoreactivity in the gracile nucleus and fasciculus in old mice. Neurosci Lett 106: 258-260. doi: 10.1016/0304-3940(89)90173-0
|
| [26] |
Yamazaki K, Moriya H, Ichihara N, et al. (1993) Substance P-immunoreactive astrocytes in gracile sensory nervous tract of spinal cord in gracile axonal dystrophy mutant mouse. Mol Chem Neuropathol 20: 1-20. doi: 10.1007/BF03160066
|
| [27] |
Matsuda T, Maeda M, Morishima Y, et al. (1985) Dystrophic axons in the nucleus gracilis of the normal rat containing cholecystokinin-like immunoreactivity: Light-and electron-microscopic observations. Acta Neuropathol 65: 224-234. doi: 10.1007/BF00687002
|
| [28] |
Li T, Chen X, Zhang C, et al. (2019) An update on reactive astrocytes in chronic pain. J Neuroinflam 16: 140. doi: 10.1186/s12974-019-1524-2
|
| [29] |
Yamazaki K, Moriya H, Wakabayashi T, et al. (1990) Glial fibrillary acidic protein-like immunoreactivity in the central nervous system of GAD (gracile axonal dystrophy) mice. Biomed Res 11: 145-149. doi: 10.2220/biomedres.11.145
|
| [30] |
Yamazaki K, Kobayashi A, Kumazawa A, et al. (1991) Axonal degeneration in the central nervous system of gracile axonal dystrophy (GAD) mice progresses like in human spinocerebellar ataxias. Biomed Res 12: 143-148. doi: 10.2220/biomedres.12.143
|
| [31] |
Dascil N, Kniewallner KM, Obermair GJ, et al. (2015) L-type calcium channel blockers and substance P induce angiogenesis of cortical vessels associated with beta-amyloid plaques in Alzheimer mouse model. Neurobiol Aging 36: 1333-1341. doi: 10.1016/j.neurobiolaging.2014.12.027
|
| [32] |
Chen XY, Du YF, Chen F (2019) Neuropeptides exert neuroprotective effects in Alzheimer's disease. Front Mol Neurosci 11: 1-19. doi: 10.3389/fnagi.2019.00001
|
| [33] |
Noguchi K, Kawai Y, Fukuoka T, et al. (1995) Substance P induced by peripheral nerve injury in primary afferent sensory neurons and its effect on dorsal column nucleus neurons. J Neurosci 15: 7633-7643. doi: 10.1523/JNEUROSCI.15-11-07633.1995
|
| [34] |
Severini C, Zona C (2006) Tachykinins and excitotoxicity in cerebellar granule cells. Cerebellum 5: 232. doi: 10.1080/14734220600673295
|
| [35] | Pieri M, Amadoro G, Carunchio I, et al. (2010) SP protect cerebellar granule cells against β-amyloid-induced apoptosis by down-regulation and reduced activity of Kv4 potassium channels. Neopharma 58: 268-276. |
| [36] |
Marolda R, Ciotti MT, Matrone C, et al. (2012) Substance P activates ADAM9 mRNA expression and inducesα-secretase-mediated amyloid precursor protein cleavage. Neurophamacol 62: 1954-1963. doi: 10.1016/j.neuropharm.2011.12.025
|
| [37] |
Kowall NW, Beal MF, Busciglio J, et al. (1991) An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance P. Proc Natl Acad Sci USA 88: 7247-7251. doi: 10.1073/pnas.88.16.7247
|
| [38] |
Campolongo P, Ratano P, Ciotti MT, et al. (2013) Systemic administration of Substance P recovers β amyloid-cognitive deficits in rat: Involvement of Kv potassium channels. Plos One 8: 1-11. doi: 10.1371/journal.pone.0078036
|
| [39] |
Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6: 487-498. doi: 10.1016/0896-6273(91)90052-2
|
| [40] |
Yamaguchi H, Nakazato Y, Hirai S, et al. (1990) Immunoelectron microscopic localization of amyloid β protein in the diffuse plaques of Alzheimer-type dementia. Brain Res 508: 320-324. doi: 10.1016/0006-8993(90)90416-9
|
| [41] |
Ichihara N, Wu J, Chui DH, et al. (1995) Axonal degeneration promotes abnormal accumulation of amyloid β-protein in ascending gracile tract of gracile axonal dystrophy (GAD) mouse. Brain Res 695: 173-178. doi: 10.1016/0006-8993(95)00729-A
|
| [42] | Ichihara N, Asari M, Kikuchi T (1997) Accumulation of amyloid β-protein in gracile tract of the gracile axonal dystrophy (GAD) mouse. J Jpn Vet Neurol 4: 3-11. |
| [43] | Ohgami T, Kitamoto T, Weidmann A, et al. (1991) Alzheimer's amyloid precursor protein-positive degenerative neurites exist even within kuru plaques not specific to Alzheimer's disease. Am J Pathol 139: 1245-1250. |
| [44] |
Kawarabayashi T, Shoji M, Yamaguchi H, et al. (1993) Amyloid β protein precursor accumulates in swollen neurites throughout rat brain with aging. Neurosci Lett 153: 73-76. doi: 10.1016/0304-3940(93)90080-5
|
| [45] |
Koo EH, Sisodia SS, Archer DR, et al. (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 87: 1561-1565. doi: 10.1073/pnas.87.4.1561
|
| [46] |
Kawarabayashi T, Shoji M, Yamaguchi H, et al. (1991) Amyloid β/A4protein precursor is widely distributed in both the central and peripheral nervous system of the mouse. Brain Res 552: 1-7. doi: 10.1016/0006-8993(91)90651-B
|
| [47] |
Otsuka N, Tomonaga M, Ikeda K (1991) Rapid appearance of β-amyloid precursor protein immunoreactivity in damaged axons and reactive grail cells in rat brain following needle stab injury. Brain Res 568: 335-338. doi: 10.1016/0006-8993(91)91422-W
|
| [48] |
Siman R, Card JP, Nelson RB, et al. (1989) Expression of β-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3: 275-285. doi: 10.1016/0896-6273(89)90252-3
|
| [49] |
Nagele RG, Wegiel J, Vengataman V, et al. (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer's disease. Nourol Aging 25: 663-674. doi: 10.1016/j.neurobiolaging.2004.01.007
|
| [50] |
Shea TB, Beermann ML, Honda T, et al. (1994) Secretion of amyloid precursor protein and laminin by cultured astrocytes is influenced by culture condition. J Neurosci Res 37: 197-207. doi: 10.1002/jnr.490370205
|
| [51] |
Willis M, Hutter-paier B, Wietzorrek G, et al. (2007) Localization and expression of substance P in transgenic mice overexpressing human APP751 with the London (V7171) and Swedish (K670M/N671L) mutations. Brain Res 1143: 199-207. doi: 10.1016/j.brainres.2007.01.080
|
| [52] | Martin LJ, Pardo CA, Cork LC, et al. (1994) Synaptic pathology and glia responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral-cortex. Am J Pathol 6: 1358-1381. |
| [53] |
Araujo DM, Cotman CW (1992) β-amyloid stimulation glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer's disease. Brain Res 569: 141-145. doi: 10.1016/0006-8993(92)90380-R
|
| [54] |
Rechsteiner M (1987) Ubiquitin-mediated pathways for intracellular proteolysis. Ann Rev Cell Biol 3: 1-30. doi: 10.1146/annurev.cb.03.110187.000245
|
| [55] | Manetto V, Abduk-Karim FW, Perry G, et al. (1989) Selective presence of ubiquitin in intracellular inclusions. Am J Pathol 134: 505-513. |
| [56] | Clechanover A, Schwartz A (1994) The ubiquitin-mediated proteolytic pathway: mechanisms of recognition of the proteolytic substrate and involvement in the degradation of native cellular proteins. FASEB J 8: 183-191. |
| [57] |
Mazurkiewicz J (1991) Ubiquitin deposits are present in spinal motor neurons in all stages of the disease in the motor neuron degeneration (Mnd) mutant of the mouse. Neurosci Lett 128: 182-186. doi: 10.1016/0304-3940(91)90256-S
|
| [58] | Wu J, Ichihara N, Chui DH, et al. (1995) Ubiquitin immunoreactivity in the central nervous system of gracile axonal dystrophy (GAD) mouse. Brain Nerve 47: 881-885. |
| [59] | Wu J, Ichihara N, Chui DH, et al. (1996) Abnormal ubiquitination of dystrophic axons in central nervous system of gracile axonal dystrophy (GAD) mutant mouse. Alzheimer's Res 2: 163-168. |
| [60] |
Saigoh K, Wang YL, Suh JG, et al. (1999) Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nature Genet 23: 47-51. doi: 10.1038/12647
|
| [61] |
Osaka H, Wang YL, Takada K, et al. (2003) Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Human Mol Genet 12: 1945-1958. doi: 10.1093/hmg/ddg211
|
| [62] |
Setsuie R, Wada K (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51: 105-111. doi: 10.1016/j.neuint.2007.05.007
|
| [63] |
Choi J, Levey AI, Weintraub ST, et al. (2004) Oxidative modification and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem 279: 13256-13264. doi: 10.1074/jbc.M314124200
|
| [64] |
Leroy E, Boyer R, Auburger G, et al. (1998) The ubiquitin pathway in Parkinson's disease. Nature 395: 451-452. doi: 10.1038/26652
|
| [65] |
Setsuie R, Wang YL, Mochizuki H, et al. (2007) Dopaminergic neuronal loss in transgenic mice expressing the Parkinson's disease-associated UCH-L1 I93M mutant. Neurochem Int 50: 119-129. doi: 10.1016/j.neuint.2006.07.015
|
| [66] |
Wang YL, Takeda A, Osaka H, et al. (2004) Accumulation of β- and γ-synucleins in the ubiquitin carboxy-terminal hydrolase L1-deficient gad mouse. Brain Res 1019: 1-9. doi: 10.1016/j.brainres.2004.05.023
|
| [67] |
Cartier AE, Djakovic SN, Saleihi A, et al. (2009) Regulation of synaptic structure by ubiquitin c-terminal hydrolase L1. J Neurosci 29: 7858-7868. doi: 10.1523/JNEUROSCI.1817-09.2009
|
| [68] |
Jara JH, Genç B, Cox CA, et al. (2015) Corticospinal motor neurons are susceptible to increased ER stress and Display profound degeneration in the absence of UCH-L1 function. Cerebral Cortex 25: 4259-4272. doi: 10.1093/cercor/bhu318
|
| [69] |
Sakurai M, Sekiguchi M, Zushida K, et al. (2008) Reduction in memory in passive avoidance learning, exploratory behavior and synaptic plasticity in mice with a spontaneous deletion in the ubiquitin C-terminal hydrolase L1 gene. Eur J Neurosci 27: 691-701. doi: 10.1111/j.1460-9568.2008.06047.x
|
| [70] |
Bilguvar K, Tyagi NK, Ozkara C, et al. (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A 110: 3489-3494. doi: 10.1073/pnas.1222732110
|
Figures(3)

Tateki Kikuchi. Circular breakdown of neural networks due to loss of deubiquitinating enzyme (UCH-L1) in gracile axonal dystrophy (gad) mouse[J]. AIMS Molecular Science, 2021, 8(4): 311-324. doi: 10.3934/molsci.2021024







 DownLoad:
DownLoad: