Citation: Atsushi Tsubota, Hidenori Ichijo, Kengo Homma. Mislocalization, aggregation formation and defect in proteolysis in ALS[J]. AIMS Molecular Science, 2016, 3(2): 246-268. doi: 10.3934/molsci.2016.2.246

| [1] |
Rosen DR, Siddique T, Patterson D, et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59-62. doi: 10.1038/362059a0

|
| [2] | Sreedharan J, Blair IP, Tripathi VB, et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 249: 1668-1672. |
| [3] |
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245-256. doi: 10.1016/j.neuron.2011.09.011
|
| [4] |
Renton AE, Majounie E, Waite A, et al. (2011) A Hexanucleotide repeat expansion in C9ORF72 is the aause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257-268. doi: 10.1016/j.neuron.2011.09.010
|
| [5] |
Majounie E, Renton AE, Mok K, et al. (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11: 323-330. doi: 10.1016/S1474-4422(12)70043-1
|
| [6] |
Keller BA, Volkening K, Droppelmann CA, et al. (2012) Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol 124: 733-747. doi: 10.1007/s00401-012-1035-z
|
| [7] | Neumann M, Sampathu DM, Kwong LK, et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 130: 130-133. |
| [8] |
Arai T, Hasegawa M, Akiyama H, et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602-611. doi: 10.1016/j.bbrc.2006.10.093
|
| [9] |
Leigh PN, Anderton BH, Dodson A, et al. (1988) Ubiquitin deposits in anterior horn cells in motor neuron disease. Neurosci Lett 93: 197-203. doi: 10.1016/0304-3940(88)90081-X
|
| [10] |
Johnson JO, Mandrioli J, Benatar M, et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68: 857-864. doi: 10.1016/j.neuron.2010.11.036
|
| [11] |
Deng H-X, Chen W, Hong S-T, et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477: 211-215. doi: 10.1038/nature10353
|
| [12] |
Maruyama H, Morino H, Ito H, et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223-226. doi: 10.1038/nature08971
|
| [13] | ALSoD (Amyotrophic Lateral Sclerosis Online Genetics Database). Available from: http://alsod.iop.kcl.ac.uk/ |
| [14] |
Cleveland DW, Laing N, Hurse PV, et al. (1995) Toxic mutants in Charcot’s sclerosis. Nature 378: 342-343. doi: 10.1038/378342a0
|
| [15] | Bruijn LI, Houseweart MK, Kato S, et al. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281: 1851-1854. |
| [16] |
Hayward LJ, Rodriguez JA, Kim JW, et al. (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem 277: 15923-15931. doi: 10.1074/jbc.M112087200
|
| [17] |
Reaume AG, Elliott JL, Hoffman EK, et al. (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13: 43-47. doi: 10.1038/ng0596-43
|
| [18] |
Fujisawa T, Homma K, Yamaguchi N, et al. (2012) A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann Neurol 72: 739-49. doi: 10.1002/ana.23668
|
| [19] |
Urushitani M, Ezzi SA, Julien JP (2007) Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 104: 2495-2500. doi: 10.1073/pnas.0606201104
|
| [20] |
Bosco DA, Morfini G, Karabacak NM, et al. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13: 1396-1403. doi: 10.1038/nn.2660
|
| [21] |
Watanabe M, Dykes-Hoberg M, Culotta VC, et al. (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8: 933-941. doi: 10.1006/nbdi.2001.0443
|
| [22] |
Basso M, Massignan T, Samengo G, et al. (2006) Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J Biol Chem 281: 33325-33335. doi: 10.1074/jbc.M603489200
|
| [23] |
Wang J, Xu G, Gonzales V, et al. (2002) Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis 10: 128-138. doi: 10.1006/nbdi.2002.0498
|
| [24] |
Furukawa Y, Fu R, Deng HX, et al. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci U S A 103: 7148-7153. doi: 10.1073/pnas.0602048103
|
| [25] |
Rodriguez JA, Valentine JS, Eggers DK, et al. (2002) Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human Copper/Zinc superoxide dismutase. J Biol Chem 277: 15932-15937. doi: 10.1074/jbc.M112088200
|
| [26] |
Sea K, Sohn SH, Durazo A, et al. (2015) Insights into the role of the unusual disulfide bond in Copper-Zinc superoxide dismutase. J Biol Chem 290: 2405-2418. doi: 10.1074/jbc.M114.588798
|
| [27] |
Doucette PA, Whitson LJ, Cao X, et al. (2004) Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant: Insights into the molecular basis for dimer stability. J Biol Chem 279: 54558-54566. doi: 10.1074/jbc.M409744200
|
| [28] |
Hough MA, Grossmann JG, Antonyuk SV, et al. (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A 101: 5976-5981. doi: 10.1073/pnas.0305143101
|
| [29] |
Araki K, Iemura S, Kamiya Y, et al. (2013) Ero1-α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J Cell Biol 202: 861-874. doi: 10.1083/jcb.201303027
|
| [30] |
Atkin JD, Farg MA, Turner BJ, et al. (2006) Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem 281: 30152-30165. doi: 10.1074/jbc.M603393200
|
| [31] |
Chen X, Zhang X, Li C, et al. (2013) S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J Neurochem 124: 45-58. doi: 10.1111/jnc.12046
|
| [32] |
Jeon GS, Nakamura T, Lee JS, et al. (2014) Potential effect of S-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol Neurobiol 49: 796-807. doi: 10.1007/s12035-013-8562-z
|
| [33] | Honjo Y, Kaneko S, Ito H, et al. (2011) Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2968: 1-7. |
| [34] |
Toichi K, Yamanaka K, Furukawa Y (2013) Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J Biol Chem 288: 4970-4980. doi: 10.1074/jbc.M112.414235
|
| [35] | Crow JP, Sampson JB, Zhuang Y, et al. (1997) Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem 69: 1936-1944. |
| [36] |
Lyons TJ, Liu H, Goto JJ, et al. (1996) Mutation in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc Natl Acad Sci U S A 93: 12240-12244. doi: 10.1073/pnas.93.22.12240
|
| [37] |
Homma K, Fujisawa T, Tsuburaya N, et al. (2013) SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol Cell 52: 75-86. doi: 10.1016/j.molcel.2013.08.038
|
| [38] |
Urushitani M, Sik A, Sakurai T, et al. (2006) Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9: 108-118. doi: 10.1038/nn1603
|
| [39] |
Israelson A, Ditsworth D, Sun S, et al. (2013) Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron 86: 218–232. doi: 10.1016/j.neuron.2015.02.034
|
| [40] |
Tan W, Naniche N, Bogush A, et al. (2013) Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J Neurosci 33: 11588-11598. doi: 10.1523/JNEUROSCI.5385-12.2013
|
| [41] |
Kikuchi H, Almer G, Yamashita S, et al. (2006) Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A 103: 6025-6030. doi: 10.1073/pnas.0509227103
|
| [42] |
Sun S, Sun Y, Ling SC, et al. (2015) Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A 112: E6993-E7002. doi: 10.1073/pnas.1520639112
|
| [43] |
Ito Y, Yamada M, Tanaka H, et al. (2009) Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol Dis 36: 470-476. doi: 10.1016/j.nbd.2009.08.013
|
| [44] |
Nishitoh H, Kadowaki H, Nagai A, et al. (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 22: 1451-1464. doi: 10.1101/gad.1640108
|
| [45] | Pokrishevsky E, Grad LI, Yousefi M, et al. (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7: 1-9. |
| [46] |
Dimos JT, Rodolfa KT, Niakan KK, et al. (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321: 1218-1221. doi: 10.1126/science.1158799
|
| [47] |
Chen H, Qian K, Du Z, et al. (2014) Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14: 796-809. doi: 10.1016/j.stem.2014.02.004
|
| [48] |
Kiskinis E, Sandoe J, Williams LA, et al. (2014) Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14: 781-795. doi: 10.1016/j.stem.2014.03.004
|
| [49] | Neary D, Snowden JS, Mann DM (2000) Classification and description of frontotemporal dementias. Ann N Y Acad Sci 920: 46-51. |
| [50] |
Higashi S, Iseki E, Yamamoto R, et al. (2007) Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184: 284-294. doi: 10.1016/j.brainres.2007.09.048
|
| [51] | Ou SH, Wu F, Harrich D, et al. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69: 3584-3596. |
| [52] |
Lu Y, Ferris J, Gao FB (2009) Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol Brain 2: 30. doi: 10.1186/1756-6606-2-30
|
| [53] |
Winton MJ, Igaz LM, Wong MM, et al. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283: 13302-13309. doi: 10.1074/jbc.M800342200
|
| [54] |
Kuo PH, Doudeva LG, Wang YT, et al. (2009) Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res 37: 1799-1808. doi: 10.1093/nar/gkp013
|
| [55] |
Buratti E, Brindisi A, Giombi M, et al. (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon9 splicing. J Biol Chem 280: 37572-37584. doi: 10.1074/jbc.M505557200
|
| [56] |
Kabashi E, Valdmanis PN, Dion P, et al. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40: 572-574. doi: 10.1038/ng.132
|
| [57] |
Johnson BS, McCaffery JM, Lindquist S, et al. (2008) A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A 105: 6439-6444. doi: 10.1073/pnas.0802082105
|
| [58] |
Hasegawa M, Arai T, Nonaka T, et al. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and ALS. Ann Neurol 64: 60-70. doi: 10.1002/ana.21425
|
| [59] |
Kadokura A, Yamazaki T, Kakuda S, et al. (2009) Phosphorylation-dependent TDP-43 antibody detects intraneuronal dot-like structures showing morphological characters of granulovacuolar degeneration. Neurosci Lett 463: 87-92. doi: 10.1016/j.neulet.2009.06.024
|
| [60] |
Igaz LM, Kwong LK, Chen-Plotkin A, et al. (2009) Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem 284: 8516-8524. doi: 10.1074/jbc.M809462200
|
| [61] |
Zhang YJ, Xu YF, Cook C, et al. (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106: 7607-7612. doi: 10.1073/pnas.0900688106
|
| [62] |
Arai T, Hasegawa M, Akiyama H, et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602-611. doi: 10.1016/j.bbrc.2006.10.093
|
| [63] |
Dormann D, Capell A, Carlson AM, et al. (2009) Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem 110: 1082-1094. doi: 10.1111/j.1471-4159.2009.06211.x
|
| [64] |
Wils H, Kleinberger G, Janssens J, et al. (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 107: 3858-3863. doi: 10.1073/pnas.0912417107
|
| [65] |
Giordana MT, Piccinini M, Grifoni S, et al. (2010) TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol 20: 351-360. doi: 10.1111/j.1750-3639.2009.00284.x
|
| [66] | Udan M, Baloh RH (2011) Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 5: 1-5. |
| [67] |
Nonaka T, Masuda-Suzukake M, Arai T, et al. (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4: 124-134. doi: 10.1016/j.celrep.2013.06.007
|
| [68] |
Walker AK, Soo KY, Sundaramoorthy V, et al. (2013) ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One 8: e81170. doi: 10.1371/journal.pone.0081170
|
| [69] |
Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14: 459-468. doi: 10.1038/nn.2779
|
| [70] |
Watanabe S, Kaneko K, Yamanaka K (2013) Accelerated disease onset with stabilized familial amyotrophic lateral ssclerosis (ALS)-linked mutant TDP-43 proteins. J Biol Chem 288: 3641-3654. doi: 10.1074/jbc.M112.433615
|
| [71] |
Iguchi Y, Katsuno M, Niwa J, et al. (2013) Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136: 1371-1382. doi: 10.1093/brain/awt029
|
| [72] |
Wegorzewska I, Bell S, Cairns NJ, et al. (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 106: 18809-18814. doi: 10.1073/pnas.0908767106
|
| [73] |
Feiguin F, Godena VK, Romano G, et al. (2009) Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583: 1586-1592. doi: 10.1016/j.febslet.2009.04.019
|
| [74] | Alami NH, Smith RB, Carrasco MA, et al. (2013) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81: 536-543. |
| [75] | Xia Q, Wang H, Hao Z, et al. (2015) TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J 35: 1-22. |
| [76] |
Armakola M, Higgins MJ, Figley MD, et al. (2012) Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44: 1302-1309. doi: 10.1038/ng.2434
|
| [77] |
Elden AC, Kim HJ, Hart MP, et al. (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069-1075. doi: 10.1038/nature09320
|
| [78] |
Liu-Yesucevitz L, Lin AY, Ebata A, et al. (2014) ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci 34: 4167-4174. doi: 10.1523/JNEUROSCI.2350-13.2014
|
| [79] |
Crozat A, Aman P, Mandahl N, et al. (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363: 640-644. doi: 10.1038/363640a0
|
| [80] |
Rabbits TH, Forster A, Larson R, et al. (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t (12; 16) in malignant liposarcoma. Nat Genet 4: 175-180. doi: 10.1038/ng0693-175
|
| [81] |
Vance C, Rogelj B, Hortobágyi T, et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis Type 6. Science 323: 1208-1211. doi: 10.1126/science.1165942
|
| [82] |
Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205-1208. doi: 10.1126/science.1166066
|
| [83] |
Neumann M, Rademakers R, Roeber S, et al. (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132: 2922-2931. doi: 10.1093/brain/awp214
|
| [84] | Kovar H (2011) The two faces of the FUS/EWS/TAF15 protein family. Sarcoma 2011: 1-13. |
| [85] |
Couthouis J, Hart MP, Erion R, et al. (2012) Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21: 2899-2911. doi: 10.1093/hmg/dds116
|
| [86] |
Lee BJ, Cansizoglu AE, Süel KE, et al. (2006) Rules for Nuclear Localization Sequence Recognition by Karyopherinβ2. Cell 126: 543-558. doi: 10.1016/j.cell.2006.05.049
|
| [87] |
Dormann D, Rodde R, Edbauer D, et al. (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29: 2841-2857. doi: 10.1038/emboj.2010.143
|
| [88] |
Dormann D, Madl T, Valori CF, et al. (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31: 4258-4275. doi: 10.1038/emboj.2012.261
|
| [89] |
Snowden JS, Hu Q, Rollinson S, et al. (2011) The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 122: 99-110. doi: 10.1007/s00401-011-0816-0
|
| [90] |
Urwin H, Josephs KA, Rohrer JD, et al. (2010) FUS pathology defines the majority of tau-and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120: 33-41. doi: 10.1007/s00401-010-0698-6
|
| [91] |
Brelstaff J, Lashley T, Holton JL, et al. (2011) Transportin1: a marker of FTLD-FUS. Acta Neuropathol 122: 591-600. doi: 10.1007/s00401-011-0863-6
|
| [92] |
Davidson YS, Robinson AC, Hu Q, et al. (2013) Nuclear carrier and RNA-binding proteins in frontotemporal lobar degeneration associated with fused in sarcoma (FUS) pathological changes. Neuropathol Appl Neurobiol 39: 157-165. doi: 10.1111/j.1365-2990.2012.01274.x
|
| [93] |
Kuroda M, Sok J, Webb L, et al. (2000) Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J 19: 453-462. doi: 10.1093/emboj/19.3.453
|
| [94] |
Hicks GG, Singh N, Nashabi A, et al. (2000) Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 24: 175-179. doi: 10.1038/72842
|
| [95] |
Kabashi E, Bercier V, Lissouba A, et al. (2011) FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 7: e1002214. doi: 10.1371/journal.pgen.1002214
|
| [96] |
Mitchell JC, McGoldrick P, Vance C, et al. (2013) Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol 125: 273-288. doi: 10.1007/s00401-012-1043-z
|
| [97] |
Levine TP, Daniels RD, Gatta AT, et al. (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29: 499-503. doi: 10.1093/bioinformatics/bts725
|
| [98] |
Farg MA, Sundaramoorthy V, Sultana JM, et al. (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23: 3579-3595. doi: 10.1093/hmg/ddu068
|
| [99] |
Donnelly CJ, Zhang PW, Pham JT, et al. (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415-428. doi: 10.1016/j.neuron.2013.10.015
|
| [100] |
Therrien M, Rouleau GA, Dion PA, et al. (2013) Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 8: 1-10. doi: 10.1371/journal.pone.0083450
|
| [101] | Ciura S, Lattante S, Le Ber I, et al. (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 74: 180-187. |
| [102] |
Koppers M, Blokhuis AM, Westeneng H-J, et al. (2015) C9orf72 ablation in mice does not cause motor neuron degenerateon or motor deficit. Ann Neurol 78: 426-438. doi: 10.1002/ana.24453
|
| [103] | Ranum LPW, Cooper TA (2006) RNA-mediated neuromascular disorders. Annu Rev Neurosci 29: 259-77. |
| [104] |
Xu Z, Poidevin M, Li X, et al. (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110: 7778-7783. doi: 10.1073/pnas.1219643110
|
| [105] | Haeusler AR, Donnelly CJ, Periz G, et al. (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195-200. |
| [106] | Zu T, Gibbens B, Doty NS, et al. (2010) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108: 260-265. |
| [107] |
Mori K, Weng S, Arzberger T, et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339: 1335-1339. doi: 10.1126/science.1232927
|
| [108] |
Mori K, Lammich S, Mackenzie IR, et al. (2013) hnRNP A3 bind to GGGGCC repeats and is a constituent of p62-positive/TDP-43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta. Neuropathol 125: 413-423. doi: 10.1007/s00401-013-1088-7
|
| [109] |
Ash PEA, Bieniek KF, Gendron TF, et al. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639-646. doi: 10.1016/j.neuron.2013.02.004
|
| [110] |
Zhang YJ, Jansen-West K, Xu YF, et al. (2014) Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128: 505-524. doi: 10.1007/s00401-014-1336-5
|
| [111] |
Mizielinska S, Grönke S, Niccoli T, et al. (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345: 1192-1195. doi: 10.1126/science.1256800
|
| [112] |
Tran H, Almeida S, Moore J, et al. (2015) Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron 87: 1207-1214 doi: 10.1016/j.neuron.2015.09.015
|
| [113] | Chew J, Gendron TF, Prudencio M, et al. (2015) C9ORF72 repeat expansion in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348: 2-6. |
| [114] |
O’Rourke JG, Bogdanik L, Muhammad AKMG, et al. (2015) C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 88: 892-901. doi: 10.1016/j.neuron.2015.10.027
|
| [115] |
Zhang K, Donnelly CJ, Haeusler AR, et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56-61. doi: 10.1038/nature14973
|
| [116] |
Freibaum BD, Lu Y, Lopez-gonzalez R, et al. (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129-133. doi: 10.1038/nature14974
|
| [117] |
Jovičić A, Mertens J, Boeynaems S, et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226-1229. doi: 10.1038/nn.4085
|
| [118] |
Rezaie T, Child A, Hitchings R, et al. (2002) Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295: 1077-1079. doi: 10.1126/science.1066901
|
| [119] |
Zhu G, Wu CJ, Zhao Y, et al. (2007) Optineurin negatively regulates TNFα- induced NF-κB activation by competing with NEMO for ubiquitinated RIP. Curr Biol 17: 1438-1443. doi: 10.1016/j.cub.2007.07.041
|
| [120] | Wild P, Farhan H, McEwan DG, et al. (2011) Phosphorylation of the autophagy receptor Optineurin restricts salmonella growth. Science 333: 228-233. |
| [121] |
Sahlender DA, Roberts RC, Arden SD, et al. (2005) Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol 169: 285-295. doi: 10.1083/jcb.200501162
|
| [122] |
Wild P, Farhan H, McEwan DG, et al. (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228-233. doi: 10.1126/science.1205405
|
| [123] | Deng HX, Bigio EH, Zhai H, et al. (2012) Differential involvement of Optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch Neurol 68: 1057-1061. |
| [124] |
Ito H, Fujita K, Nakamura M, et al. (2011) Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol 121: 555-557. doi: 10.1007/s00401-011-0809-z
|
| [125] | Williams KL, Warraich ST, Yang S, et al. (2012) UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 33: 2527.e3-10. |
| [126] |
Seok Ko H, Uehara T, Tsuruma K, et al. (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett 566: 110-114. doi: 10.1016/j.febslet.2004.04.031
|
| [127] |
Ritson GP, Custer SK, Freibaum BD, et al. (2010) TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30: 7729-7739. doi: 10.1523/JNEUROSCI.5894-09.2010
|
| [128] |
Ye Y, Shibata Y, Yun C, et al. (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429: 841-847. doi: 10.1038/nature02656
|
| [129] |
Ye Y, Shibata Y, Kikkert M, et al. (2005) Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A 102: 14132-14138. doi: 10.1073/pnas.0505006102
|
| [130] |
Song C, Wang Q, Li CH (2003) ATPase Activity of p97-Valosin-containing Protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem 278: 3648-3655. doi: 10.1074/jbc.M208422200
|
| [131] |
Carvalho P, Stanley AM, Rapoport TA (2010) Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143: 579-591. doi: 10.1016/j.cell.2010.10.028
|
| [132] |
Bilican B, Serio A, Barmada SJ, et al. (2012) Mutant induced plu- ripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A 109: 5803-5808. doi: 10.1073/pnas.1202922109
|
| [133] |
Liu X, Chen J, Liu W, et al. (2015) The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics 16: 223-231. doi: 10.1007/s10048-015-0448-y
|
| [134] |
Mackenzie IR, Bigio EH, Ince PG, et al. (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61: 427-434. doi: 10.1002/ana.21147
|
| [135] |
Yamanaka K, Chun SJ, Boillee S, et al. (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11: 251-253. doi: 10.1038/nn2047
|
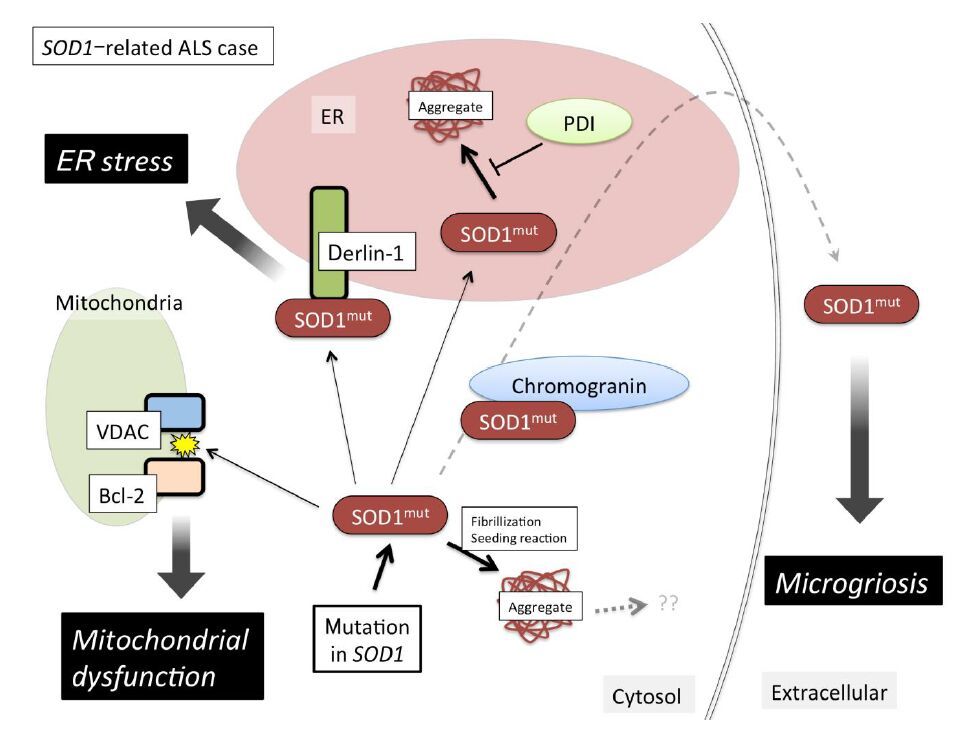
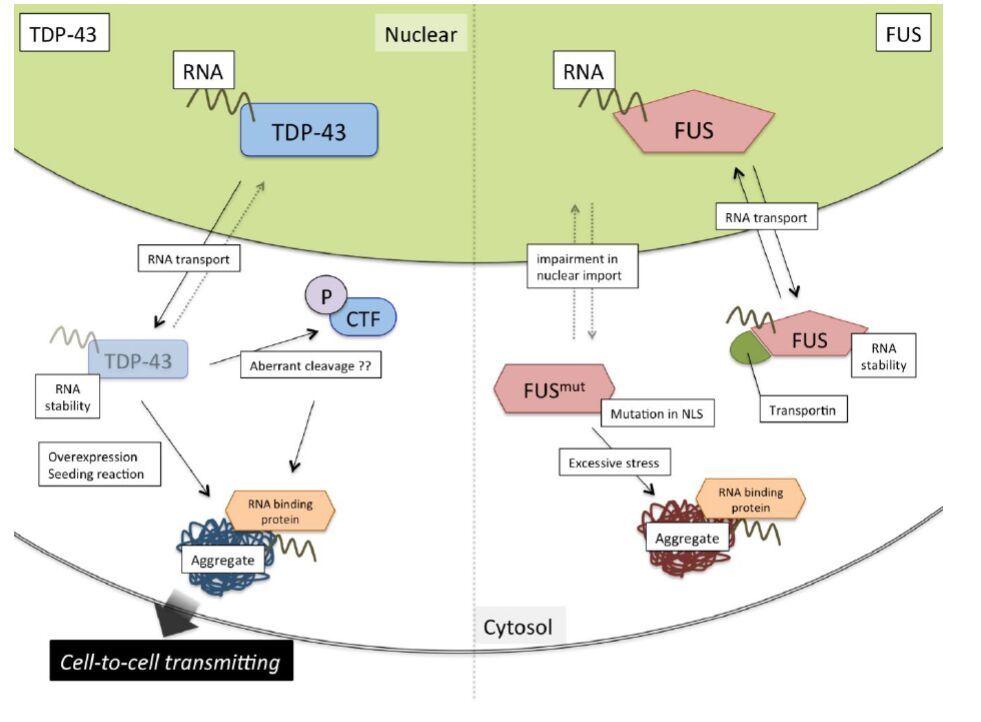
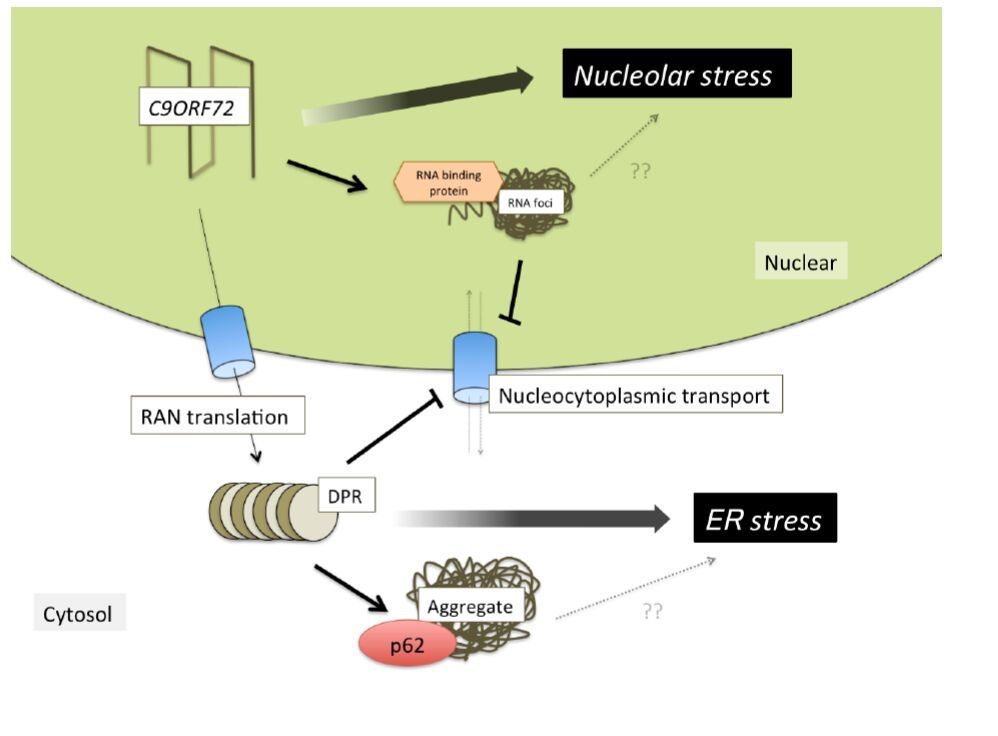
Figures(3)

Atsushi Tsubota, Hidenori Ichijo, Kengo Homma. Mislocalization, aggregation formation and defect in proteolysis in ALS[J]. AIMS Molecular Science, 2016, 3(2): 246-268. doi: 10.3934/molsci.2016.2.246







 DownLoad:
DownLoad: