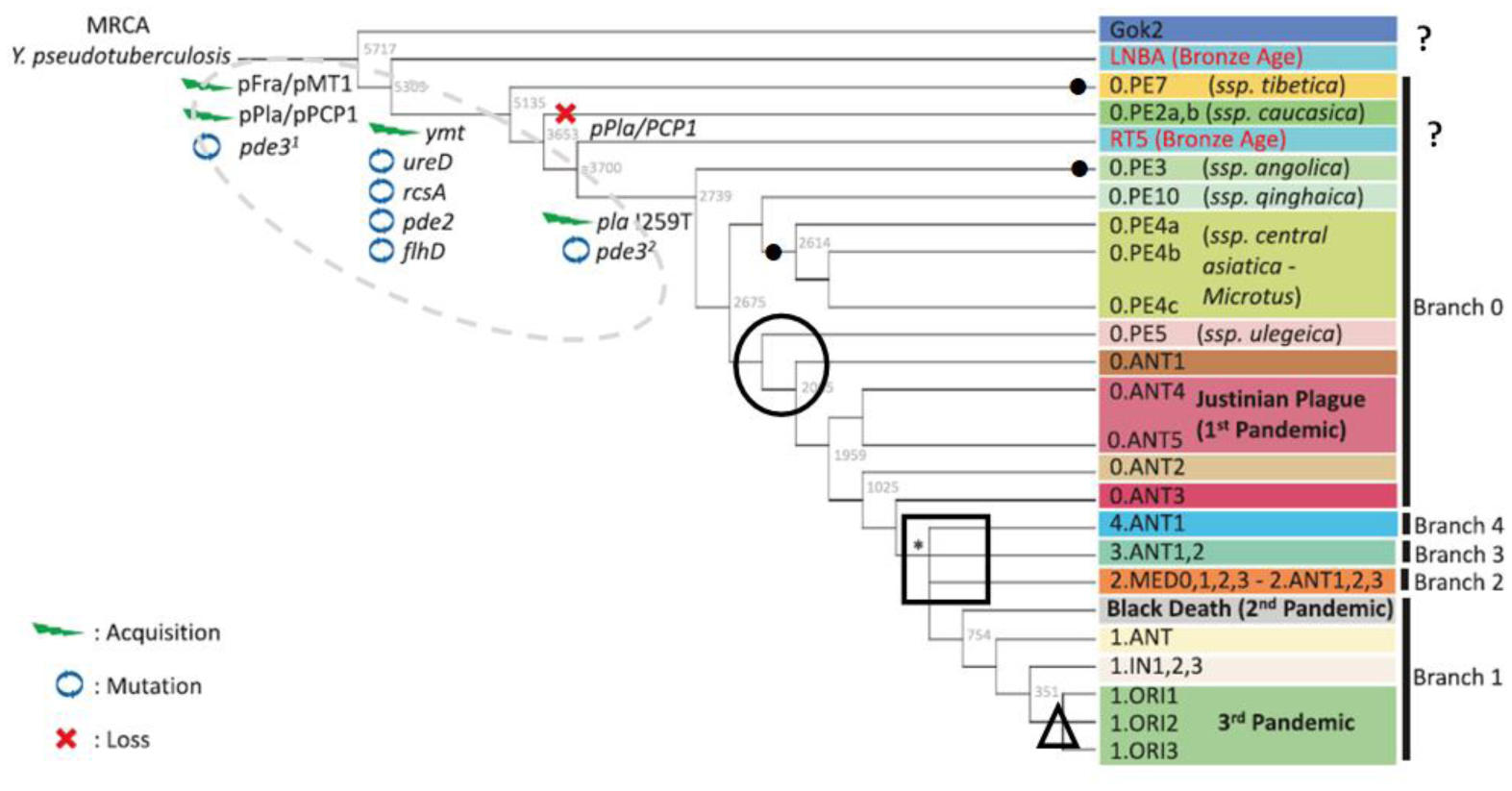
Two approaches are applied to studies of the phylogeny of the plague microbe Yersinia pestis, i.e., the reconstruction of its history: Molecular genetic (MG) and ecological (ECO). The MG approach dominates. Phylogenies created with MG and ECO methods are not congruent. MG conclusions contradict the known facts and patterns of ecology, biogeography, paleontology, etc. We discuss some obvious contradictions and inconsistencies and suggest that real phylogenies of the plague microbe can be constructed only on the basis of the integration of MG and ECO approaches.
Citation: Victor V. Suntsov. Molecular phylogenies of the plague microbe Yersinia pestis: an environmental assessment[J]. AIMS Microbiology, 2023, 9(4): 712-723. doi: 10.3934/microbiol.2023036
Two approaches are applied to studies of the phylogeny of the plague microbe Yersinia pestis, i.e., the reconstruction of its history: Molecular genetic (MG) and ecological (ECO). The MG approach dominates. Phylogenies created with MG and ECO methods are not congruent. MG conclusions contradict the known facts and patterns of ecology, biogeography, paleontology, etc. We discuss some obvious contradictions and inconsistencies and suggest that real phylogenies of the plague microbe can be constructed only on the basis of the integration of MG and ECO approaches.

| [1] | Wu LT, Chum JWH, Pollitzer R, et al. (1936) Plague: a manual for medical and public health workers. Shanghai: Mercury Press 547. |
| [2] | Devignat R (1951) Varietes de l'espese Pasteurella pestis. Nouvelle hypothese // Bull. WHO 4: 242-263. |
| [3] | Rall, Yu M (1965) Natural focility and epizootology of plague. Moscow: Medicine 363. |
| [4] |
Achtman M, Morelli G, Zhu P, et al. (2004) Microevolution and history of the plague bacillus. Yersinia pestis. PNAS 51: 17837-17842. https://doi10.1073pnas.0408026101 
|
| [5] |
Morelli G, Song Y, Mazzoni CJ, et al. (2010) Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet 12: 1140-1145. https://doi:10.1038/ng.705
|
| [6] |
Skurnik M, Peippo A, Ervela E (2000) Characterization of the O-antigen gene cluster of Yersinia pseudotuberculosis and the cryptic O-antigen gene cluster of Yersinia pestis shows that the plague bacillus is most closely related to and has evolved from Y. pseudotuberculosis serotype O:1b. Mol Microbiol 2: 316-330. https://doi.org/10.1046/j.1365-2958.2000.01993.x
|
| [7] |
Eppinger M, Rosovitz MJ, Fricke WF, et al. (2007) The complete genome sequence of Yersinia pseudotuberculosis IP31758, the causative agent of Far East scarlet-like fever. PLoS Genet 3: e142. https://doi:10.1371/journal.pgen.0030142
|
| [8] | Somov GP, Pokrovskii VI, Besednova NN, et al. (2001) Pseudotuberculosis. Moscow: Medicine 254. |
| [9] |
Fukushima H, Matsuda Y, Seki R, et al. (2001) Geographical heterogeneity between Far Eastern and Western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains. J Clin Microbiol 10: 3541-35479. https://doi:10.1128/JCM.39.10.3541–3547.2001
|
| [10] |
Suntsov VV (2022) Climate Changes in Central Asia as a Prerequisite and Trigger of Plague Microbe (Yersinia pestis) Speciation. Contemp Probl Ecol 15: 373-382. https://doi:10.1134/S1995425522040102
|
| [11] |
Suntsov VV (2021) Host aspect of territorial expansion of the plague microbe Yersinia pestis from the populations of the Tarbagan Marmot (Marmota sibirica). Biol Bull 48: 211-223. https://doi:10.1134/S1062359021080288
|
| [12] |
Suntsov VV (2022) Polytopic speciation of the plague microbe Yersinia pestis as the cause of the Trichotomy in the geographic populations of the Mongolian Tarbagan Marmot (Marmota sibirica). Biol Bull Rev 12: 504-515. https://doi:10.1134/S2079086422050085
|
| [13] |
Cui Y, Yu C, Yan Y, et al. (2013) Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. PNAS 2: 577-582. https://doi/10.1073/pnas.1205750110
|
| [14] |
Kutyrev VV, Eroshenko GA, Motin VL, et al. (2018) Phylogeny and classification of Yersinia pestis through the lens of strains from the plague foci of commonwealth of independent states. Front Microbiol 9: 1106. https://doi:10.3389/fmicb.2018.01106
|
| [15] |
Demeure CE, Dussurget O, Fiol GM, et al. (2019) Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Genes Immun 5: 357-370. https://doi.org/10.1038/s41435-019-0065-0
|
| [16] |
Valtuena AA, Neumann GU, Spyrou MA, et al. (2022) Stone age Yersinia pestis genomes shed light on the early evolution, diversity, and ecology of plague. PNAS Genet 119: e2116722119. https://doi.org/10.1073/pnas.2116722119
|
| [17] | Eroshenko GA, Kukleva LM, Kutyrev VV (2022) Historical and modern classifications of the plague agent. Probl Particularly Dangerous Infect 4: 14-22. https://doi:10.21055/0370-1069-2022-4-14-22 |
| [18] |
Stenseth NC, Taoc Y, Zhang C, et al. (2022) No evidence for persistent natural plague reservoirs in historical and modern Europe. PNAS 119: e2209816119. https://doi.org/10.1073/pnas.2209816119
|
| [19] |
Sun YC, Jarrett CO, Bosio CF, Hinnebusch BJ (2014) Retracing the evolutionary path that led to flea-borne transmission of Yersinia pestis. Cell Host Microbe 15: 578-586. https://doi.org/10.1016/j.chom.2014.04.003
|
| [20] |
Hinnebusch BJ, Chouikha I, Sun YC (2016) Ecological opportunity, evolution, and the emergence of flea-borne plague. Infect Immun 84: 1932-1940. https://doi:10.1128/IAI.00188-16
|
| [21] |
Wren BW (2003) The Yersinia—a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1: 55-64. https://doi:10.1038/nrmicro730
|
| [22] |
Eppinger M, Worsham PL, Nikolich MP, et al. (2010) Genome sequence of the deep-rooted Yersinia pestis strain angola reveals new insights into the evolution and pangenome of the plague bacterium. J Bacteriol 192: 1685-1699. https://doi:10.1128/JB.01518-09
|
| [23] | Platonov ME, Evseeva VV, Dentovskaya SV, et al. (2013) Molecular typing of Yersinia pestis. Mol Genetic Microbiol Virusol 2: 3-12. https://doi:10.1111/j.1469-0691.2008.01953.x |
| [24] |
Kislichkina AA, Platonov ME, Vagaiskaya AS, et al. (2019) Rational taxonomy of Yersinia pestis. Mol Genetic Microbiol Virusol 2: 76-82. https://doi.org/10.17116/molgen20193702176
|
| [25] |
Suntsov VV (2023) Parallelism in speciation and intraspecific diversification of the plague microbe Yersinia pestis. Biol Bull 50: 103-109. https://doi:10.1134/S1062359023010120
|
| [26] |
Suntsov VV (2022) Phylogenesis of the plague microbe Yersinia pestis: the uniqueness of the evolutionary model. Herald Rus Acad Sci 92: 609-616. https://doi:10.1134/S1019331622050057
|
| [27] | Suntsov VV (2022) Ecological scenario of the plague microbe Yersinia pestis speciation underlying adequate molecular evolutionary model. Rus Jour Infect Immun 12: 809-818. https://doi:10.15789/2220-7619-ESO-1955 |
| [28] |
Suntsov VV (2018) Quantum speciation of Yersinia pestis plague microbe in a heteroimmune environment: in the populations of hibernating tarbagan marmots (Marmota sibirica). Contemp Probl Ecol 11: 343-354. https://doi:10.1134/S199542551804008X
|
| [29] |
Suntsov VV (2019) Origin of the plague: prospects of ecological-molecular-genetic synthesis. Her Rus Acad Sci 89: 271-278. https://doi.org/10.1134/S1019331619010118
|

Figures(2)

Victor V. Suntsov. Molecular phylogenies of the plague microbe Yersinia pestis: an environmental assessment[J]. AIMS Microbiology, 2023, 9(4): 712-723. doi: 10.3934/microbiol.2023036







 DownLoad:
DownLoad: