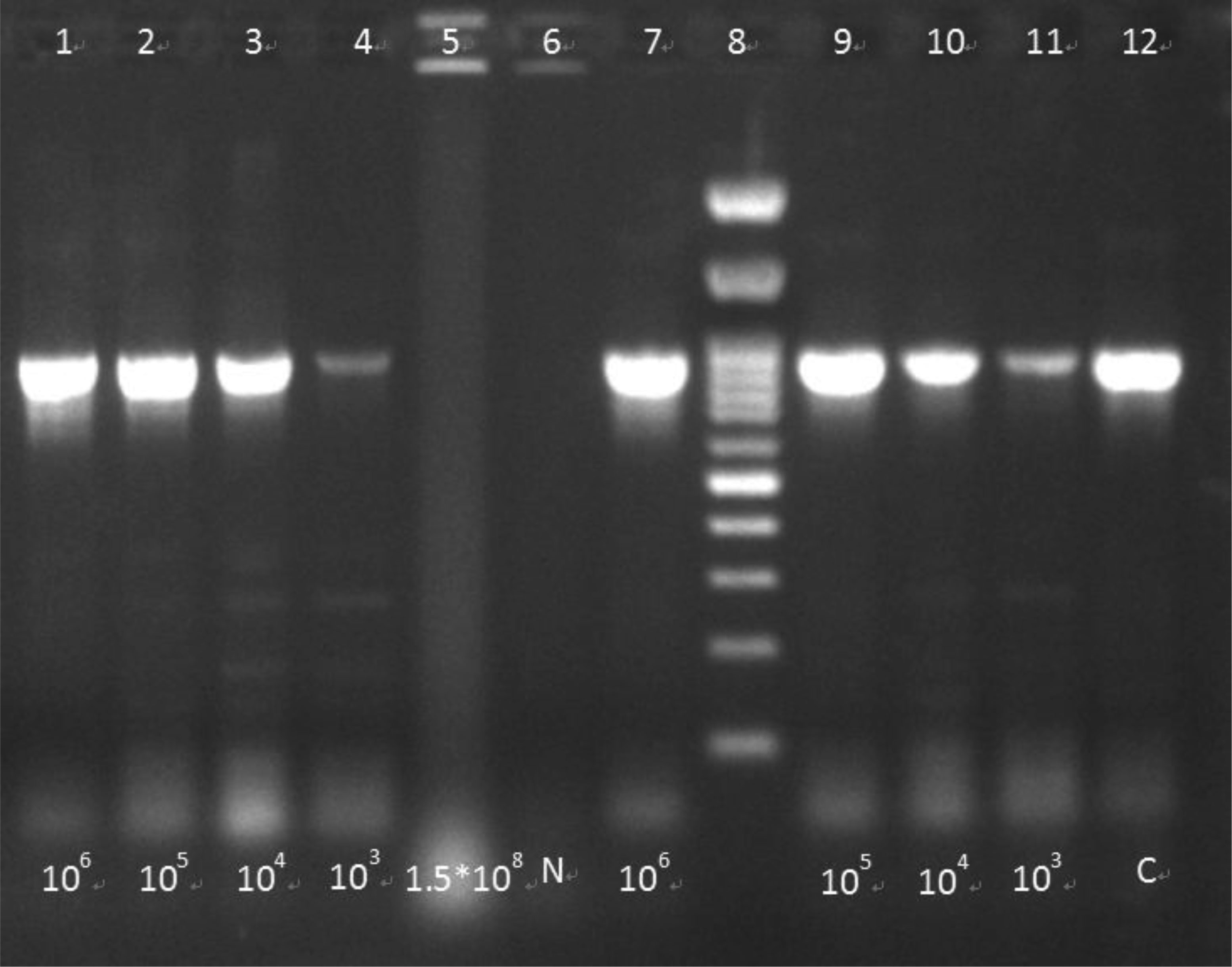
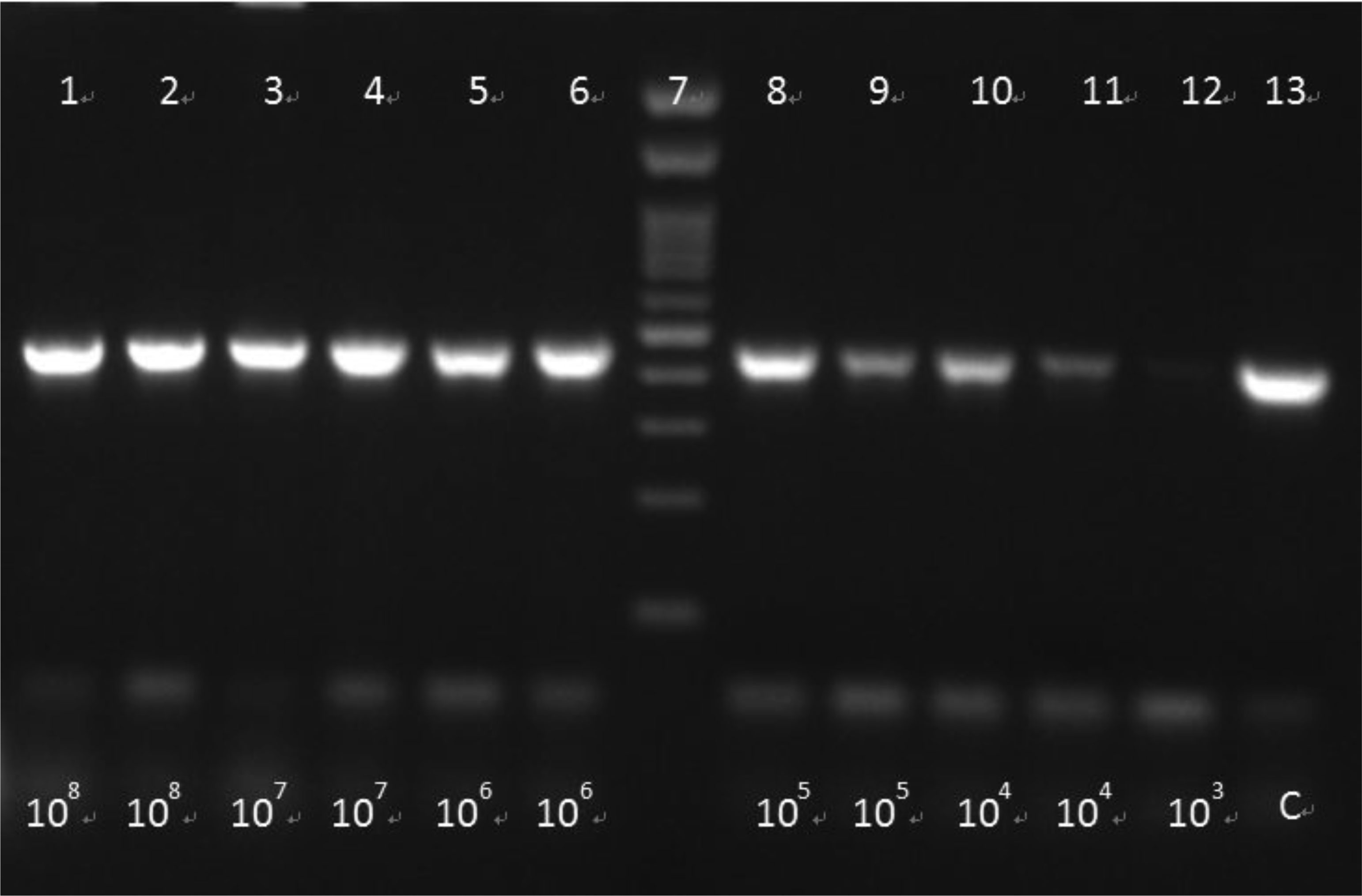
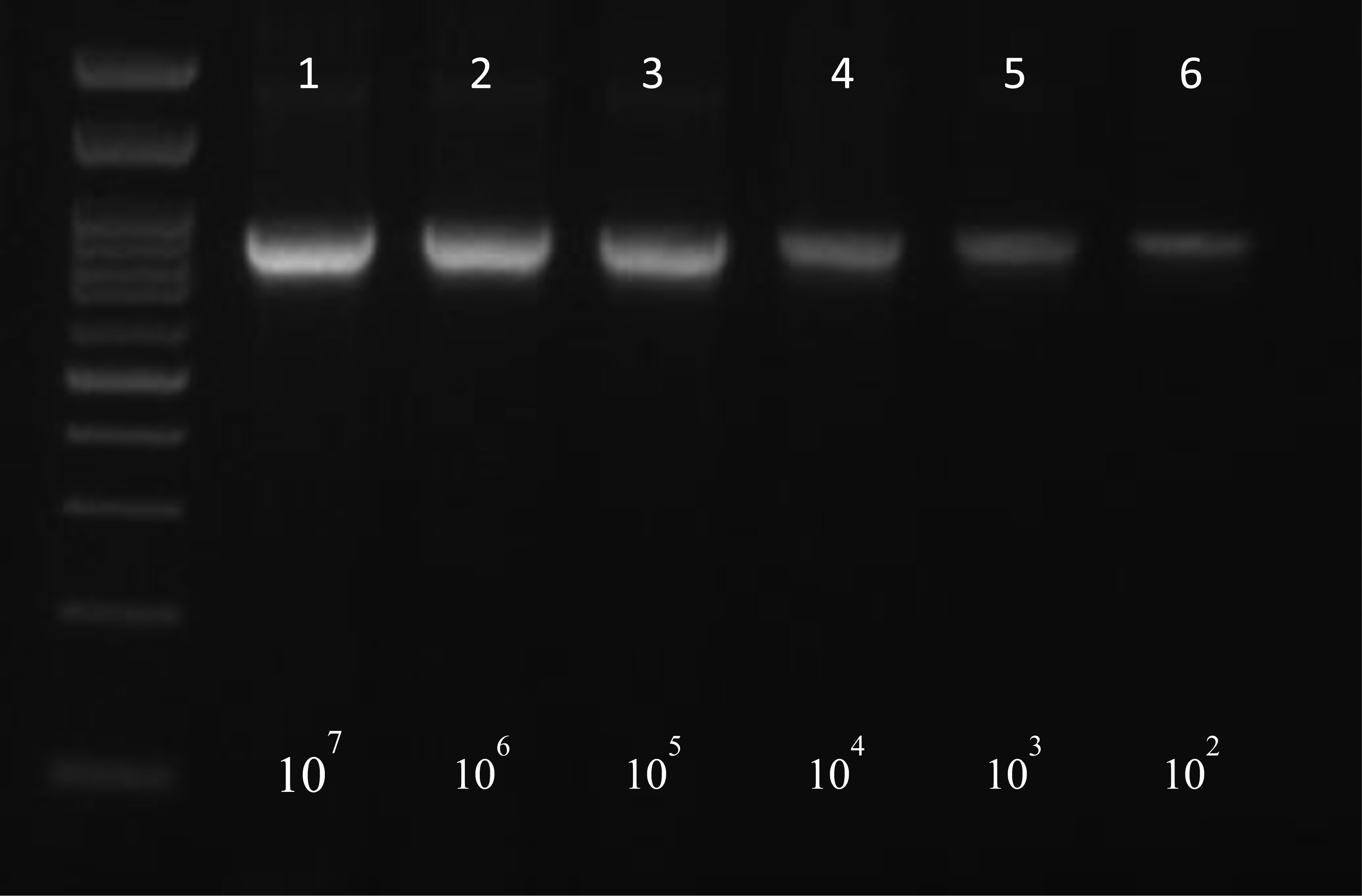
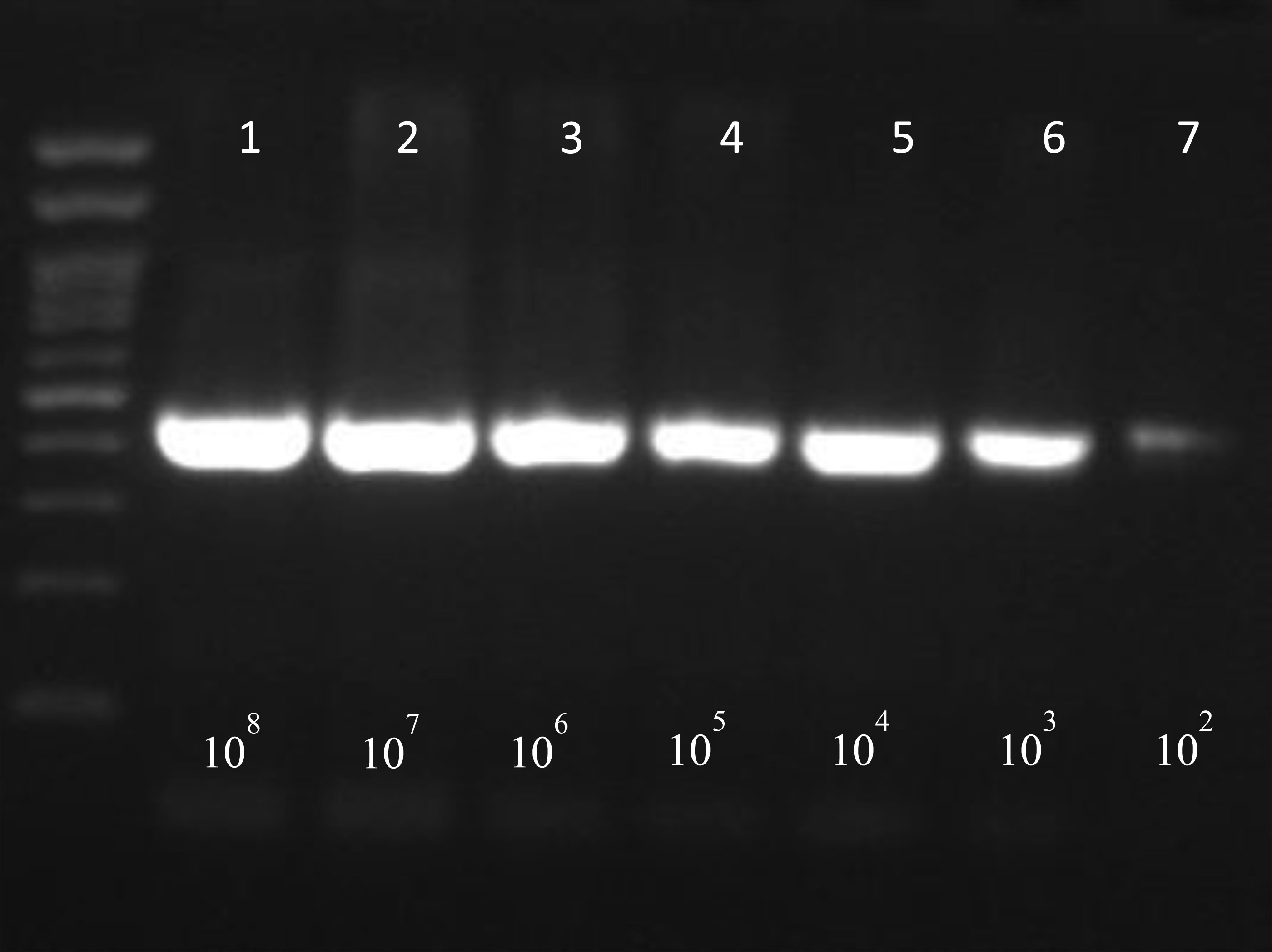
Foodborne infections continue to plague Europe. Food safety monitoring is in crisis as the existing techniques for detecting pathogens do not keep up with the global rising of food production and consumption. Thus, the development of innovative techniques for detecting and identifying pathogenic bacteria has become critical. The aim of the present study was firstly to develop an innovative simple and low cost method of extracting bacterial DNA from contaminated food and water samples with Salmonella enteric(a) subsp. enteric(a) serovar Typhimurium and Listeria monocytogenes and its comparison with two commercial DNA extraction kits (Qiagen, Macherey-Nagel). Finally, pathogens' detection using two molecular techniques (PCR-electrophoresis, LAMP), in order to evaluate the best combination of DNA extraction and identification based on their sensitivity, cost, rapidity and simplicity. Considering the above criteria, among them, best was proved an in-house bacterial DNA extraction method, based on the chloroform-isoamyl alcohol protocol, with certain modifications. This technique showed statistically similar results in terms of sensitivity, compared to the commercial kits, while at the same time maintained high rapidity and much lower cost. Lastly, between the molecular techniques, LAMP was found more promising considering its simplicity, high rapidity and sensitivity. Conclusively, the in-house DNA extraction method along with the LAMP technique, was proven to be the best among the presented combinations.
Citation: Spyridon Andreas Papatheodorou, Panagiotis Halvatsiotis, Dimitra Houhoula. A comparison of different DNA extraction methods and molecular techniques for the detection and identification of foodborne pathogens[J]. AIMS Microbiology, 2021, 7(3): 304-319. doi: 10.3934/microbiol.2021019
Foodborne infections continue to plague Europe. Food safety monitoring is in crisis as the existing techniques for detecting pathogens do not keep up with the global rising of food production and consumption. Thus, the development of innovative techniques for detecting and identifying pathogenic bacteria has become critical. The aim of the present study was firstly to develop an innovative simple and low cost method of extracting bacterial DNA from contaminated food and water samples with Salmonella enteric(a) subsp. enteric(a) serovar Typhimurium and Listeria monocytogenes and its comparison with two commercial DNA extraction kits (Qiagen, Macherey-Nagel). Finally, pathogens' detection using two molecular techniques (PCR-electrophoresis, LAMP), in order to evaluate the best combination of DNA extraction and identification based on their sensitivity, cost, rapidity and simplicity. Considering the above criteria, among them, best was proved an in-house bacterial DNA extraction method, based on the chloroform-isoamyl alcohol protocol, with certain modifications. This technique showed statistically similar results in terms of sensitivity, compared to the commercial kits, while at the same time maintained high rapidity and much lower cost. Lastly, between the molecular techniques, LAMP was found more promising considering its simplicity, high rapidity and sensitivity. Conclusively, the in-house DNA extraction method along with the LAMP technique, was proven to be the best among the presented combinations.

| [1] | WHO The Burden of Foodborne Diseases in the WHO European Region (2017) . |
| [2] | Baraketi A, Salmieri S, Lacroix M (2018) Foodborne pathogens detection: persevering worldwide challenge amina. Biosensing Technologies for the Detection of Pathogens-A Prospective Way For Rapid Analysis Croatia: InTech publishing, 53-72. |
| [3] |
Sayad A, Ibrahim F, Uddin MS, et al. (2018) A microdevice for rapid, monoplex and colorimetric detection of foodborne pathogens using a centrifugal microfluidic platform. Biosens Bioel 100: 96-104. doi: 10.1016/j.bios.2017.08.060

|
| [4] | EFSA and ECDC (2016) The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2015. EFSA Journal 14: 4634. |
| [5] | Marquis H, Drevets DA, Bronze M S, et al. (2016) Pathogenesis of Listeria monocytogenes in Humans. Human Emerging and Re-emerging Infections: Viral and Parasitic Infections USA: Willey-Blackwell, 749-772. |
| [6] |
Radoshevich L, Cossart P (2017) Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol 16: 32-46. doi: 10.1038/nrmicro.2017.126
|
| [7] |
Santos RL, Tsolis RM, Adams LG, et al. (2003) Pathogenesis of Salmonella-induced enteritis. Braz J Med Biol Res 36: 3-12. doi: 10.1590/S0100-879X2003000100002
|
| [8] |
Zhang S, Kingsley RA, Santos RL, et al. (2003) Molecular pathogenesis of Salmonella enterica serotype typhimurium-induced diarrhea. Inf Immun 71: 1-12. doi: 10.1128/IAI.71.1.1-12.2003
|
| [9] |
Rajapaksha P, Elbourne A (2018) A review of methods for the detection of pathogenic microorganisms. Analyst 144: 396-411. doi: 10.1039/C8AN01488D
|
| [10] |
Liu Y, Cao Y, Wang T, et al. (2019) Detection of 12 common food-borne bacterial pathogens by TaqMan Real-Time PCR using a single set of reaction conditions. Front Microbiol 10: 1-9. doi: 10.3389/fmicb.2019.00001
|
| [11] |
Schirone M, Visciano P, Tofalo R, et al. (2019) Editorial: foodborne pathogens: hygiene and safety. Front Microbiol 10: 1-4. doi: 10.3389/fmicb.2019.01974
|
| [12] |
Charlermroj R, Makornwattana M, Phuengwas S, et al. (2019) DNA-based bead array technology for simultaneous identification of eleven foodborne pathogens in chicken meat. Food Cont 101: 81-88. doi: 10.1016/j.foodcont.2019.02.014
|
| [13] |
Rodrigues C, Desai N, Fernandes H (2016) Clinical Diagnosis in Resource-Limited Settings. Clinl Microbiol News 38: 51-56. doi: 10.1016/j.clinmicnews.2016.03.004
|
| [14] | Klančnik A, Kovač M (2012) PCR in Food Analysis. Polymerase Chain Reaction China: Tech Publishing, 195-220. |
| [15] |
Kaneko H, Kawana T, Fukushima E, et al. (2007) Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J Biochem Biophys Meth 70: 499-501. doi: 10.1016/j.jbbm.2006.08.008
|
| [16] |
Lee JE, Kang LH, Kim JO, et al. (2019) Rapid detection of carbapenemase-producing Enterobacteriaceae (CPE) using a simple DNA extraction method and Loop-mediated isothermal amplification (LAMP). J Microbiol Meth 167: 105746. doi: 10.1016/j.mimet.2019.105746
|
| [17] | Law JW, Mutalib NA, Chan K, et al. (2015) Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front Microbiol 5: 1-20. |
| [18] |
Mateus-Barros E, Meneghine AK, Bagatini IL, et al. (2019) Comparison of two DNA extraction methods widely used in aquatic microbial ecology. J Microbiol Meth 159: 12-17. doi: 10.1016/j.mimet.2019.02.005
|
| [19] |
Tanner NA, Zhang Y, Evans TC (2015) Visual detection of isothermal nucleic acidamplification using pH-sensitive dyes. Biotechn 58: 59-68. doi: 10.2144/000114253
|
| [20] | Anju P, Latha C, Sunil B, et al. (2014) Detection of Salmonella and Yersinia spp . in uncooked retail chicken meat in Kerala by multiplex PCR. Int J Cur Microbiol Ap Sci 3: 1028-1034. |
| [21] |
Khalil ZK (2017) A study of isolation and identification of Campylobacter species, Listeria monocytogenes, E. coli O157:H7 and Salmonella species from raw chicken carcasses and beef meat by multiplex polymer chain reaction in Baghdad city. Inte J Adv Res 5: 900-907. doi: 10.21474/IJAR01/5134
|
| [22] |
Srisawat M, Panbangred W (2015) Efficient and specific detection of Salmonella in food samples using a stn-Based Loop-Mediated isothermal amplification method. BioMed Res Int 176: 1-7. doi: 10.1155/2015/356401
|
| [23] |
Tang MJ, Zhou S, Zhang XY, et al. (2011) Rapid and sensitive detection of Listeria monocytogenes by Loop-Mediated isothermal amplification. Cur Microbiol 63: 511-516. doi: 10.1007/s00284-011-0013-3
|
| [24] |
Psi A, Dovas CI, Banos G (2010) A comparison of six methods for genomic DNA extraction suitable for PCR-based genotyping applications using bovine milk samples. Mol Cel Prob 24: 93-98. doi: 10.1016/j.mcp.2009.11.001
|
| [25] |
Khan M, Wang R, Li B, et al. (2018) Comparative Evaluation of the LAMP assay and PCR-Based assays for the rpid dtection of Alternaria solani. Front Microbiol 9: 2089. doi: 10.3389/fmicb.2018.02089
|
| [26] |
Wang X, Seo DJ, Lee MH, et al. (2014) Comparison of cnventional PCR, mltiplex PCR, and Loop-Mediated iothermal aplification asays for rpid dtection of Arcobacter secies. J Clin Microbiol 52: 557. doi: 10.1128/JCM.02883-13
|
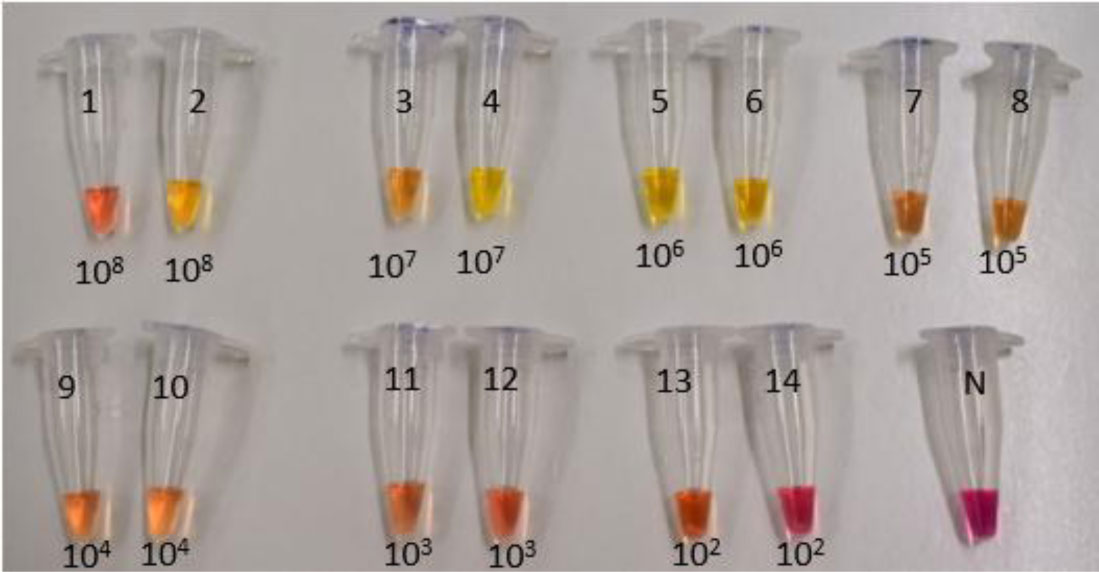
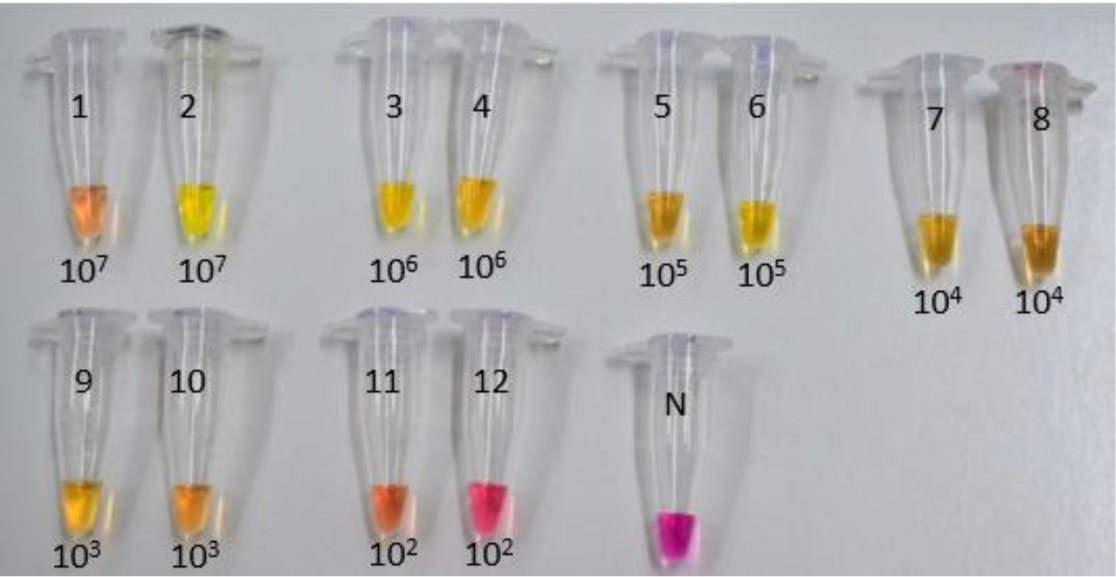

Figures(7) / Tables(3)

Spyridon Andreas Papatheodorou, Panagiotis Halvatsiotis, Dimitra Houhoula. A comparison of different DNA extraction methods and molecular techniques for the detection and identification of foodborne pathogens[J]. AIMS Microbiology, 2021, 7(3): 304-319. doi: 10.3934/microbiol.2021019







 DownLoad:
DownLoad: