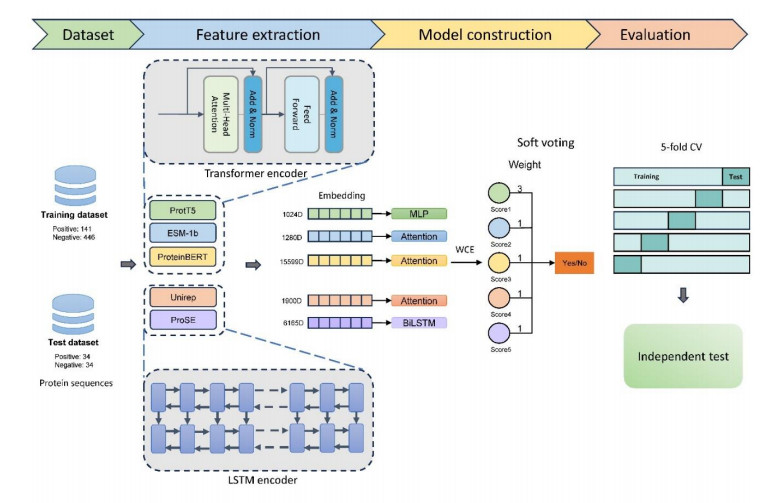
Non-classical secreted proteins (NCSPs) refer to a group of proteins that are located in the extracellular environment despite the absence of signal peptides and motifs. They usually play different roles in intercellular communication. Therefore, the accurate prediction of NCSPs is a critical step to understanding in depth their associated secretion mechanisms. Since the experimental recognition of NCSPs is often costly and time-consuming, computational methods are desired. In this study, we proposed an ensemble learning framework, termed NCSP-PLM, for the identification of NCSPs by extracting feature embeddings from pre-trained protein language models (PLMs) as input to several fine-tuned deep learning models. First, we compared the performance of nine PLM embeddings by training three neural networks: Multi-layer perceptron (MLP), attention mechanism and bidirectional long short-term memory network (BiLSTM) and selected the best network model for each PLM embedding. Then, four models were excluded due to their below-average accuracies, and the remaining five models were integrated to perform the prediction of NCSPs based on the weighted voting. Finally, the 5-fold cross validation and the independent test were conducted to evaluate the performance of NCSP-PLM on the benchmark datasets. Based on the same independent dataset, the sensitivity and specificity of NCSP-PLM were 91.18% and 97.06%, respectively. Particularly, the overall accuracy of our model achieved 94.12%, which was 7~16% higher than that of the existing state-of-the-art predictors. It indicated that NCSP-PLM could serve as a useful tool for the annotation of NCSPs.
Citation: Taigang Liu, Chen Song, Chunhua Wang. NCSP-PLM: An ensemble learning framework for predicting non-classical secreted proteins based on protein language models and deep learning[J]. Mathematical Biosciences and Engineering, 2024, 21(1): 1472-1488. doi: 10.3934/mbe.2024063
Non-classical secreted proteins (NCSPs) refer to a group of proteins that are located in the extracellular environment despite the absence of signal peptides and motifs. They usually play different roles in intercellular communication. Therefore, the accurate prediction of NCSPs is a critical step to understanding in depth their associated secretion mechanisms. Since the experimental recognition of NCSPs is often costly and time-consuming, computational methods are desired. In this study, we proposed an ensemble learning framework, termed NCSP-PLM, for the identification of NCSPs by extracting feature embeddings from pre-trained protein language models (PLMs) as input to several fine-tuned deep learning models. First, we compared the performance of nine PLM embeddings by training three neural networks: Multi-layer perceptron (MLP), attention mechanism and bidirectional long short-term memory network (BiLSTM) and selected the best network model for each PLM embedding. Then, four models were excluded due to their below-average accuracies, and the remaining five models were integrated to perform the prediction of NCSPs based on the weighted voting. Finally, the 5-fold cross validation and the independent test were conducted to evaluate the performance of NCSP-PLM on the benchmark datasets. Based on the same independent dataset, the sensitivity and specificity of NCSP-PLM were 91.18% and 97.06%, respectively. Particularly, the overall accuracy of our model achieved 94.12%, which was 7~16% higher than that of the existing state-of-the-art predictors. It indicated that NCSP-PLM could serve as a useful tool for the annotation of NCSPs.

| [1] |
M. Zhang, L. Liu, X. Lin, Y. Wang, Y. Li, Q. Guo, et al., A translocation pathway for vesicle-mediated unconventional protein secretion, Cell, 181 (2020), 637–652. https://doi.org/10.1016/j.cell.2020.03.031 doi: 10.1016/j.cell.2020.03.031

|
| [2] |
Q. Kang, D. Zhang, Principle and potential applications of the non-classical protein secretory pathway in bacteria, Appl. Microbiol. Biotechnol., 104 (2020), 953–965. https://doi.org/10.1007/s00253-019-10285-4 doi: 10.1007/s00253-019-10285-4
|
| [3] |
M. Jacopo, Unconventional protein secretion (UPS): Role in important diseases, Mol. Biomed., 4 (2023), 2. https://doi.org/10.1186/s43556-022-00113-z doi: 10.1186/s43556-022-00113-z
|
| [4] |
P. Broz, Unconventional protein secretion by gasdermin pores, Semin. Immunol., 69 (2023), 101811. https://doi.org/10.1016/j.smim.2023.101811 doi: 10.1016/j.smim.2023.101811
|
| [5] |
G. Poschmann, J. Bahr, J. Schrader, I. Stejerean-Todoran, I. Bogeski, K. Stuehler, Secretomics-a key to a comprehensive picture of unconventional protein secretion, Front. Cell. Dev. Biol., 10 (2022), 828027. https://doi.org/10.3389/fcell.2022.878027 doi: 10.3389/fcell.2022.878027
|
| [6] |
W. Dai, J. Li, Q. Li, J. Cai, J. Su, C. Stubenrauch, et al., PncsHub: A platform for annotating and analyzing non-classically secreted proteins in Gram-positive bacteria, Nucleic Acids Res., 50 (2022), D848–D857. https://doi.org/10.1093/nar/gkab814 doi: 10.1093/nar/gkab814
|
| [7] |
J. D. Bendtsen, L. J. Jensen, N. Blom, G. von Heijne, S. Brunak, Feature-based prediction of non-classical and leaderless protein secretion, Protein Eng. Des. Sel., 17 (2004), 349–356. https://doi.org/10.1093/protein/gzh037 doi: 10.1093/protein/gzh037
|
| [8] |
L. Yu, Y. Guo, Z. Zhang, Y. Li, M. Li, G. Li, et al., SecretP: A new method for predicting mammalian secreted proteins, Peptides, 31 (2010), 574–578. https://doi.org/10.1016/j.peptides.2009.12.026 doi: 10.1016/j.peptides.2009.12.026
|
| [9] |
D. Restrepo-Montoya, C. Pino, L. F. Nino, M. E. Patarroyo, M. A. Patarroyo, NClassG+: A classifier for non-classically secreted Gram-positive bacterial proteins, BMC Bioinf., 12 (2011), 21. https://doi.org/10.1186/1471-2105-12-21 doi: 10.1186/1471-2105-12-21
|
| [10] |
Y. Zhang, S. Yu, R. Xie, J. Li, A. Leier, T.T. Marquez-Lago, et al., PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins, Bioinf., 36 (2020), 704–712. https://doi.org/10.1093/bioinformatics/btz629 doi: 10.1093/bioinformatics/btz629
|
| [11] |
C. Wang, J. Wu, L. Xu, Q. Zou, NonClasGP-Pred: Robust and efficient prediction of non-classically secreted proteins by integrating subset-specific optimal models of imbalanced data, Microb. Genom., 6 (2020), mgen000483. https://doi.org/10.1099/mgen.0.000483 doi: 10.1099/mgen.0.000483
|
| [12] |
X. Wang, F. Li, J. Xu, J. Rong, G. I. Webb, Z. Ge, et al., ASPIRER: A new computational approach for identifying non-classical secreted proteins based on deep learning, Brief. Bioinf., 23 (2022), bbac031. https://doi.org/10.1093/bib/bbac031 doi: 10.1093/bib/bbac031
|
| [13] |
T. T. Do, T. H. Nguyen-Vo, H. T. Pham, Q. H. Trinh, B. P. Nguyen, iNSP-GCAAP: Identifying nonclassical secreted proteins using global composition of amino acid properties, Proteomics, 23 (2023), e2100134. https://doi.org/10.1002/pmic.202100134 doi: 10.1002/pmic.202100134
|
| [14] |
H. Zulfiqar, Z. Guo, B. K. Grace-Mercure, Z. Y. Zhang, H. Gao, H. Lin, et al., Empirical comparison and recent advances of computational prediction of hormone binding proteins using machine learning methods, Comput. Struct. Biotechnol. J., 21 (2023), 2253–2261. https://doi.org/10.1016/j.csbj.2023.03.024 doi: 10.1016/j.csbj.2023.03.024
|
| [15] |
F. Y. Dao, M. L. Liu, W. Su, H. Lv, Z. Y. Zhang, H. Lin, et al., AcrPred: A hybrid optimization with enumerated machine learning algorithm to predict anti-CRISPR proteins, Int. J. Biol. Macromol., 228 (2023), 706–714. https://doi.org/10.1016/j.ijbiomac.2022.12.250 doi: 10.1016/j.ijbiomac.2022.12.250
|
| [16] |
S. F. Altschul, T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang, W. Miller, et al., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs, Nucleic Acids Res., 25 (1997), 3389–3402. https://doi.org/10.1093/nar/25.17.3389 doi: 10.1093/nar/25.17.3389
|
| [17] |
E. Asgari, M. R. K. Mofrad, Continuous distributed representation of biological sequences for deep proteomics and genomics, PloS One, 10 (2015), e0141287. https://doi.org/10.1371/journal.pone.0141287 doi: 10.1371/journal.pone.0141287
|
| [18] |
M. Heinzinger, A. Elnaggar, Y. Wang, C. Dallago, D. Nechaev, F. Matthes, et al., Modeling aspects of the language of life through transfer-learning protein sequences, BMC Bioinf., 20 (2019), 723. https://doi.org/10.1186/s12859-019-3220-8 doi: 10.1186/s12859-019-3220-8
|
| [19] |
T. Bepler, B. Berger, Learning the protein language: Evolution, structure, and function, Cell Syst., 12 (2021), 654–669. https://doi.org/10.1016/j.cels.2021.05.017 doi: 10.1016/j.cels.2021.05.017
|
| [20] |
E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi, G. M. Church, Unified rational protein engineering with sequence-based deep representation learning, Nat. Methods, 16 (2019), 1315–1322. https://doi.org/10.1038/s41592-019-0598-1 doi: 10.1038/s41592-019-0598-1
|
| [21] | R. Rao, N. Bhattacharya, N. Thomas, Y. Duan, X. Chen, J. Canny, et al., Evaluating protein transfer learning with TAPE, in 33rd Conference on Neural Information Processing Systems (NeurIPS), 32 (2019), 9689–9701. |
| [22] |
A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin, J. Liu, et al., Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proc. Natl. Acad. Sci. U. S. A., 118 (2021), e2016239118. https://doi.org/10.1073/pnas.2016239118 doi: 10.1073/pnas.2016239118
|
| [23] |
A. Elnaggar, M. Heinzinger, C. Dallago, G. Rehawi, Y. Wang, L. Jones, et al., ProtTrans: Toward understanding the language of life through self-supervised learning, IEEE Trans. Pattern Anal. Mach. Intell., 44 (2022), 7112–7127. https://doi.org/10.1109/tpami.2021.3095381 doi: 10.1109/tpami.2021.3095381
|
| [24] |
N. Brandes, D. Ofer, Y. Peleg, N. Rappoport, M. Linial, ProteinBERT: A universal deep-learning model of protein sequence and function, Bioinformatics, 38 (2022), 2102–2110. https://doi.org/10.1093/bioinformatics/btac020 doi: 10.1093/bioinformatics/btac020
|
| [25] |
V. Thumuluri, J. J. A. Armenteros, A. R. Johansen, H. Nielsen, O. Winther, DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models, Nucleic Acids Res., 50 (2022), W228–W234. https://doi.org/10.1093/nar/gkac278 doi: 10.1093/nar/gkac278
|
| [26] |
L. Wang, C. Huang, M. Wang, Z. Xue, Y. Wang, NeuroPred-PLM: An interpretable and robust model for neuropeptide prediction by protein language model, Brief. Bioinf., 24 (2023), bbad077. https://doi.org/10.1093/bib/bbad077 doi: 10.1093/bib/bbad077
|
| [27] |
Z. Du, X. Ding, W. Hsu, A. Munir, Y. Xu, Y. Li, pLM4ACE: A protein language model based predictor for antihypertensive peptide screening, Food Chem., 431 (2024), 137162–137162. https://doi.org/10.1016/j.foodchem.2023.137162 doi: 10.1016/j.foodchem.2023.137162
|
| [28] |
A. Villegas-Morcillo, A. M. Gomez, V. Sanchez, An analysis of protein language model embeddings for fold prediction, Brief. Bioinf., 23 (2022), bbac142. https://doi.org/10.1093/bib/bbac142 doi: 10.1093/bib/bbac142
|
| [29] |
P. Pratyush, S. Pokharel, H. Saigo, D. B. Kc, pLMSNOSite: An ensemble-based approach for predicting protein S-nitrosylation sites by integrating supervised word embedding and embedding from pre-trained protein language model, BMC Bioinf., 24 (2023), 41. https://doi.org/10.1186/s12859-023-05164-9 doi: 10.1186/s12859-023-05164-9
|
| [30] |
X. Wang, Z. Ding, R. Wang, X. Lin, Deepro-Glu: Combination of convolutional neural network and Bi-LSTM models using ProtBert and handcrafted features to identify lysine glutarylation sites, Brief. Bioinf., 24 (2023), bbac631. https://doi.org/10.1093/bib/bbac631 doi: 10.1093/bib/bbac631
|
| [31] |
E. Fenoy, A.A. Edera, G. Stegmayer, Transfer learning in proteins: Evaluating novel protein learned representations for bioinformatics tasks, Brief. Bioinf., 23 (2022), bbac232. https://doi.org/10.1093/bib/bbac232 doi: 10.1093/bib/bbac232
|
| [32] |
X. Peng, X. Wang, Y. Guo, Z. Ge, F. Li, X. Gao, et al., RBP-TSTL is a two-stage transfer learning framework for genome-scale prediction of RNA-binding proteins, Brief. Bioinf., 23 (2022), bbac215. https://doi.org/10.1093/bib/bbac215 doi: 10.1093/bib/bbac215
|
| [33] |
B. Boeckmann, A. Bairoch, R. Apweiler, M.C. Blatter, A. Estreicher, E. Gasteiger, et al., The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003, Nucleic Acids Res., 31 (2003), 365–370. https://doi.org/10.1093/nar/gkg095 doi: 10.1093/nar/gkg095
|
| [34] |
B. E. Suzek, Y. Wang, H. Huang, P. B. McGarvey, C. H. Wu, C. UniProt, UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches, Bioinformatics, 31 (2015), 926–932. https://doi.org/10.1093/bioinformatics/btu739 doi: 10.1093/bioinformatics/btu739
|
| [35] |
A. G. Murzin, S. E. Brenner, T. Hubbard, C. Chothia, SCOP-A structural classification of proteins database for the investigation of sequences and structures, J. Mol. Biol., 247 (1995), 536–540. https://doi.org/10.1016/s0022-2836(05)80134-2 doi: 10.1016/s0022-2836(05)80134-2
|
| [36] |
R. D. Finn, A. Bateman, J. Clements, P. Coggill, R. Y. Eberhardt, S. R. Eddy, et al., Pfam: The protein families database, Nucleic Acids Res., 42 (2014), D222–D230. https://doi.org/10.1093/nar/gkt1223 doi: 10.1093/nar/gkt1223
|
| [37] |
M. Steinegger, M. Mirdita, J. Soeding, Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold, Nat. Methods, 16 (2019), 603–606. https://doi.org/10.1038/s41592-019-0437-4 doi: 10.1038/s41592-019-0437-4
|
| [38] |
N.V. Chawla, K. W. Bowyer, L. O. Hall, W.P. Kegelmeyer, SMOTE: Synthetic minority over-sampling technique, J. Artif. Intell. Res., 16 (2002), 321–357. https://doi.org/10.1613/jair.953 doi: 10.1613/jair.953
|
| [39] |
T. Y. Lin, P. Goyal, R. Girshick, K. He, P. Dollar, Focal loss for dense object detection, IEEE Trans. Pattern Anal. Mach. Intell., 42 (2020), 318–327. https://doi.org/10.1109/tpami.2018.2858826 doi: 10.1109/tpami.2018.2858826
|
| [40] | S. Jadon, Ieee, A survey of loss functions for semantic segmentation, in IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), (2020), 115–121. https://doi.org/10.1109/cibcb48159.2020.9277638 |
| [41] |
S. S. Yuan, D. Gao, X. Q. Xie, C. Y. Ma, W. Su, Z. Y. Zhang, et al., IBPred: A sequence-based predictor for identifying ion binding protein in phage, Comput. Struct. Biotechnol. J., 20 (2022), 4942–4951. https://doi.org/10.1016/j.csbj.2022.08.053 doi: 10.1016/j.csbj.2022.08.053
|
| [42] |
Y. H. Wang, Y. F. Zhang, Y. Zhang, Z. F. Gu, Z. Y. Zhang, H. Lin, et al., Identification of adaptor proteins using the ANOVA feature selection technique, Methods, 208 (2022), 42–47. https://doi.org/10.1016/j.ymeth.2022.10.008 doi: 10.1016/j.ymeth.2022.10.008
|






Figures(7) / Tables(6)

Taigang Liu, Chen Song, Chunhua Wang. NCSP-PLM: An ensemble learning framework for predicting non-classical secreted proteins based on protein language models and deep learning[J]. Mathematical Biosciences and Engineering, 2024, 21(1): 1472-1488. doi: 10.3934/mbe.2024063







 DownLoad:
DownLoad: