Citation: Janine E. Deakin, Renae Domaschenz, Pek Siew Lim, Tariq Ezaz, Sudha Rao. Comparative epigenomics: an emerging field with breakthrough potential to understand evolution of epigenetic regulation[J]. AIMS Genetics, 2014, 1(1): 34-54. doi: 10.3934/genet.2014.1.34

| [1] |
Consortium EP, Bernstein BE, Birney E, et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57-74. doi: 10.1038/nature11247

|
| [2] | Waddington CH (1942) The epigenotype. Endeavour 1: 18-20. |
| [3] |
Lee JT (2011) Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nat Rev Mol Cell Biol 12: 815-826. doi: 10.1038/nrm3231
|
| [4] |
Koerner MV, Barlow DP (2010) Genomic imprinting-an epigenetic gene-regulatory model. Curr Opin Genet Dev 20: 164-170. doi: 10.1016/j.gde.2010.01.009
|
| [5] |
Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4: 143-153. doi: 10.1038/nrc1279
|
| [6] |
Lim PS, Li J, Holloway AF, et al. (2013) Epigenetic regulation of inducible gene expression in the immune system. Immunology 139: 285-293. doi: 10.1111/imm.12100
|
| [7] |
Gomes MV, Pelosi GG (2013) Epigenetic vulnerability and the environmental influence on health. Exp Biol Med (Maywood) 238: 859-865. doi: 10.1177/1535370213490630
|
| [8] |
Richards EJ (2008) Population epigenetics. Curr Opin Genet Dev 18: 221-226. doi: 10.1016/j.gde.2008.01.014
|
| [9] | Gupta S (2013) Epigenetics posited as important for evolutionary success. Nature News. |
| [10] |
Liebl AL, Schrey AW, Richards CL, et al. (2013) Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr Comp Biol 53: 351-358. doi: 10.1093/icb/ict007
|
| [11] |
Richards CL, Schrey AW, Pigliucci M (2012) Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol Lett 15: 1016-1025. doi: 10.1111/j.1461-0248.2012.01824.x
|
| [12] | Luger K, Mader AW, Richmond RK, et al. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251-260. |
| [13] |
Hager GL, McNally JG, Misteli T (2009) Transcription dynamics. Mol Cell 35: 741-753. doi: 10.1016/j.molcel.2009.09.005
|
| [14] |
Reid G, Gallais R, Metivier R (2009) Marking time: the dynamic role of chromatin and covalent modification in transcription. Int J Biochem Cell Biol 41: 155-163. doi: 10.1016/j.biocel.2008.08.028
|
| [15] | Wolffe AP (2001) Histone genes. In: Breener S, Miller JH, editors. Encyclopedia of Genetics. San Diego & London: Academic Press. 948-952. |
| [16] |
Munshi A, Shafi G, Aliya N, et al. (2009) Histone modifications dictate specific biological readouts. J Genet Genomics 36: 75-88. doi: 10.1016/S1673-8527(08)60094-6
|
| [17] |
Peterson CL, Laniel MA (2004) Histones and histone modifications. Curr Biol 14: R546-551. doi: 10.1016/j.cub.2004.07.007
|
| [18] |
Tan M, Luo H, Lee S, et al. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146: 1016-1028. doi: 10.1016/j.cell.2011.08.008
|
| [19] |
Ho JW, Jung YL, Liu T, et al. (2014) Comparative analysis of metazoan chromatin organization. Nature 512: 449-452. doi: 10.1038/nature13415
|
| [20] |
Fuchs J, Demidov D, Houben A, et al. (2006) Chromosomal histone modification patterns--from conservation to diversity. Trends Plant Sci 11: 199-208. doi: 10.1016/j.tplants.2006.02.008
|
| [21] |
de Ruijter AJ, van Gennip AH, Caron HN, et al. (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370: 737-749. doi: 10.1042/BJ20021321
|
| [22] |
Marmorstein R, Roth SY (2001) Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev 11: 155-161. doi: 10.1016/S0959-437X(00)00173-8
|
| [23] |
Marzluff WF, Gongidi P, Woods KR, et al. (2002) The human and mouse replication-dependent histone genes. Genomics 80: 487-498. doi: 10.1006/geno.2002.6850
|
| [24] | Boulard M, Bouvet P, Kundu TK, et al. (2007) Histone variant nucleosomes: structure, function and implication in disease. Subcell Biochem 41: 71-89. |
| [25] | Talbert PB, Henikoff S (2010) Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11: 264-275. |
| [26] | Yelagandula R, Stroud H, Holec S, et al. (2014) The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 158: 98-109. |
| [27] |
Marino-Ramirez L, Kann MG, Shoemaker BA, et al. (2005) Histone structure and nucleosome stability. Expert Rev Proteomics 2: 719-729. doi: 10.1586/14789450.2.5.719
|
| [28] |
Talbert PB, Ahmad K, Almouzni G, et al. (2012) A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 5: 7. doi: 10.1186/1756-8935-5-7
|
| [29] |
Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273-304. doi: 10.1146/annurev.biochem.77.062706.153223
|
| [30] |
Smith CL, Peterson CL (2005) A conserved Swi2/Snf2 ATPase motif couples ATP hydrolysis to chromatin remodeling. Mol Cell Biol 25: 5880-5892. doi: 10.1128/MCB.25.14.5880-5892.2005
|
| [31] | Saha A, Wittmeyer J, Cairns BR (2006) Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 7: 437-447. |
| [32] |
Shen W, Xu C, Huang W, et al. (2007) Solution structure of human Brg1 bromodomain and its specific binding to acetylated histone tails. Biochemistry 46: 2100-2110. doi: 10.1021/bi0611208
|
| [33] |
Mohrmann L, Verrijzer CP (2005) Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim Biophys Acta 1681: 59-73. doi: 10.1016/j.bbaexp.2004.10.005
|
| [34] |
Bultman S, Gebuhr T, Yee D, et al. (2000) A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell 6: 1287-1295. doi: 10.1016/S1097-2765(00)00127-1
|
| [35] |
Bestor TH, Verdine GL (1994) DNA methyltransferases. Curr Opin Cell Biol 6: 380-389. doi: 10.1016/0955-0674(94)90030-2
|
| [36] |
Ooi SK, O'Donnell AH, Bestor TH (2009) Mammalian cytosine methylation at a glance. J Cell Sci 122: 2787-2791. doi: 10.1242/jcs.015123
|
| [37] |
Goll MG, Bestor TH (2005) Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481-514. doi: 10.1146/annurev.biochem.74.010904.153721
|
| [38] |
Jeltsch A (2010) Phylogeny of methylomes. Science 328: 837-838. doi: 10.1126/science.1190738
|
| [39] |
Lyko F, Foret S, Kucharski R, et al. (2010) The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol 8: e1000506. doi: 10.1371/journal.pbio.1000506
|
| [40] |
Feng S, Cokus SJ, Zhang X, et al. (2010) Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 107: 8689-8694. doi: 10.1073/pnas.1002720107
|
| [41] |
Zemach A, McDaniel IE, Silva P, et al. (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916-919. doi: 10.1126/science.1186366
|
| [42] |
Zilberman D, Gehring M, Tran RK, et al. (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39: 61-69. doi: 10.1038/ng1929
|
| [43] |
Falckenhayn C, Boerjan B, Raddatz G, et al. (2013) Characterization of genome methylation patterns in the desert locust Schistocerca gregaria. J Exp Biol 216: 1423-1429. doi: 10.1242/jeb.080754
|
| [44] |
Fneich S, Dheilly N, Adema C, et al. (2013) 5-methyl-cytosine and 5-hydroxy-methyl-cytosine in the genome of Biomphalaria glabrata, a snail intermediate host of Schistosoma mansoni. Parasit Vectors 6: 167. doi: 10.1186/1756-3305-6-167
|
| [45] |
Tahiliani M, Koh KP, Shen Y, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930-935. doi: 10.1126/science.1170116
|
| [46] |
Spruijt CG, Gnerlich F, Smits AH, et al. (2013) Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152: 1146-1159. doi: 10.1016/j.cell.2013.02.004
|
| [47] |
Almeida RD, Sottile V, Loose M, et al. (2012) Semi-quantitative immunohistochemical detection of 5-hydroxymethyl-cytosine reveals conservation of its tissue distribution between amphibians and mammals. Epigenetics 7: 137-140. doi: 10.4161/epi.7.2.18949
|
| [48] |
Almeida RD, Loose M, Sottile V, et al. (2012) 5-hydroxymethyl-cytosine enrichment of non-committed cells is not a universal feature of vertebrate development. Epigenetics 7: 383-389. doi: 10.4161/epi.19375
|
| [49] | Wojciechowski M, Rafalski D, Kucharski R, et al. (2014) Insights into DNA hydroxymethylation in the honeybee from in-depth analyses of TET dioxygenase. Open Biol 4. |
| [50] |
Khalil AM, Guttman M, Huarte M, et al. (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106: 11667-11672. doi: 10.1073/pnas.0904715106
|
| [51] |
Koziol MJ, Rinn JL (2010) RNA traffic control of chromatin complexes. Curr Opin Genet Dev 20: 142-148. doi: 10.1016/j.gde.2010.03.003
|
| [52] |
Mercer TR, Mattick JS (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20: 300-307. doi: 10.1038/nsmb.2480
|
| [53] |
Bernstein E, Allis CD (2005) RNA meets chromatin. Genes Dev 19: 1635-1655. doi: 10.1101/gad.1324305
|
| [54] |
Gendrel AV, Colot V (2005) Arabidopsis epigenetics: when RNA meets chromatin. Curr Opin Plant Biol 8: 142-147. doi: 10.1016/j.pbi.2005.01.007
|
| [55] |
Hall IM, Noma K, Grewal SI (2003) RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci U S A 100: 193-198. doi: 10.1073/pnas.232688099
|
| [56] |
Volpe TA, Kidner C, Hall IM, et al. (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297: 1833-1837. doi: 10.1126/science.1074973
|
| [57] |
Grewal SI, Moazed D (2003) Heterochromatin and epigenetic control of gene expression. Science 301: 798-802. doi: 10.1126/science.1086887
|
| [58] |
Hannon GJ (2002) RNA interference. Nature 418: 244-251. doi: 10.1038/418244a
|
| [59] |
Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431: 343-349. doi: 10.1038/nature02873
|
| [60] |
Tomari Y, Zamore PD (2005) Perspective: machines for RNAi. Genes Dev 19: 517-529. doi: 10.1101/gad.1284105
|
| [61] |
Quach H, Barreiro LB, Laval G, et al. (2009) Signatures of purifying and local positive selection in human miRNAs. Am J Hum Genet 84: 316-327. doi: 10.1016/j.ajhg.2009.01.022
|
| [62] |
Clark AM, Goldstein LD, Tevlin M, et al. (2010) The microRNA miR-124 controls gene expression in the sensory nervous system of Caenorhabditis elegans. Nucleic Acids Res 38: 3780-3793. doi: 10.1093/nar/gkq083
|
| [63] |
Aboobaker AA, Tomancak P, Patel N, et al. (2005) Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc Natl Acad Sci U S A 102: 18017-18022. doi: 10.1073/pnas.0508823102
|
| [64] |
Deo M, Yu JY, Chung KH, et al. (2006) Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn 235: 2538-2548. doi: 10.1002/dvdy.20847
|
| [65] |
Wienholds E, Kloosterman WP, Miska E, et al. (2005) MicroRNA expression in zebrafish embryonic development. Science 309: 310-311. doi: 10.1126/science.1114519
|
| [66] | Mor E, Shomron N (2013) Species-specific microRNA regulation influences phenotypic variability: perspectives on species-specific microRNA regulation. Bioessays 35: 881-888. |
| [67] | Xi QY, Xiong YY, Wang YM, et al. (2014) Genome-wide discovery of novel and conserved microRNAs in white shrimp (Litopenaeus vannamei). Mol Biol Rep [Epub ahead of print]. |
| [68] |
Jain M, Chevala VN, Garg R (2014) Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J Exp Bot 65: 5945-5958. doi: 10.1093/jxb/eru333
|
| [69] |
Cowled C, Stewart CR, Likic VA, et al. (2014) Characterisation of novel microRNAs in the Black flying fox (Pteropus alecto) by deep sequencing. BMC Genomics 15: 682. doi: 10.1186/1471-2164-15-682
|
| [70] |
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68-73. doi: 10.1093/nar/gkt1181
|
| [71] |
Disteche CM (2012) Dosage compensation of the sex chromosomes. Annu Rev Genet 46: 537-560. doi: 10.1146/annurev-genet-110711-155454
|
| [72] |
Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190: 372-373. doi: 10.1038/190372a0
|
| [73] |
Deakin JE (2013) Marsupial X chromosome inactivation: past, present and future. Aust J Zool 61: 13-23. doi: 10.1071/ZO12113
|
| [74] |
Glas R, Marshall Graves JA, Toder R, et al. (1999) Cross-species chromosome painting between human and marsupial directly demonstrates the ancient region of the mammalian X. Mamm Genome 10: 1115-1116. doi: 10.1007/s003359901174
|
| [75] |
Graves JA (1995) The evolution of mammalian sex chromosomes and the origin of sex determining genes. Philos Trans R Soc Lond B Biol Sci 350: 305-311; discussion 311-302. doi: 10.1098/rstb.1995.0166
|
| [76] |
Grant J, Mahadevaiah SK, Khil P, et al. (2012) Rsx is a metatherian RNA with Xist-like properties in X-chromosome inactivation. Nature 487: 254-258. doi: 10.1038/nature11171
|
| [77] |
Borsani G, Tonlorenzi R, Simmler MC, et al. (1991) Characterization of a murine gene expressed from the inactive X chromosome. Nature 351: 325-329. doi: 10.1038/351325a0
|
| [78] |
Heard E (2005) Delving into the diversity of facultative heterochromatin: the epigenetics of the inactive X chromosome. Curr Opin Genet Dev 15: 482-489. doi: 10.1016/j.gde.2005.08.009
|
| [79] |
Koina E, Chaumeil J, Greaves IK, et al. (2009) Specific patterns of histone marks accompany X chromosome inactivation in a marsupial. Chromosome Res 17: 115-126. doi: 10.1007/s10577-009-9020-7
|
| [80] |
Rens W, Wallduck MS, Lovell FL, et al. (2010) Epigenetic modifications on X chromosomes in marsupial and monotreme mammals and implications for evolution of dosage compensation. Proc Natl Acad Sci U S A 107: 17657-17662. doi: 10.1073/pnas.0910322107
|
| [81] |
Zakharova IS, Shevchenko AI, Shilov AG, et al. (2011) Histone H3 trimethylation at lysine 9 marks the inactive metaphase X chromosome in the marsupial Monodelphis domestica. Chromosoma 120: 177-183. doi: 10.1007/s00412-010-0300-y
|
| [82] |
Chaumeil J, Waters PD, Koina E, et al. (2011) Evolution from XIST-independent to XIST-controlled X-chromosome inactivation: epigenetic modifications in distantly related mammals. PLoS One 6: e19040. doi: 10.1371/journal.pone.0019040
|
| [83] |
Mahadevaiah SK, Royo H, VandeBerg JL, et al. (2009) Key features of the X inactivation process are conserved between marsupials and eutherians. Curr Biol 19: 1478-1484. doi: 10.1016/j.cub.2009.07.041
|
| [84] |
Plath K, Fang J, Mlynarczyk-Evans SK, et al. (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131-135. doi: 10.1126/science.1084274
|
| [85] |
Zhao J, Sun BK, Erwin JA, et al. (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750-756. doi: 10.1126/science.1163045
|
| [86] |
Costanzi C, Pehrson JR (1998) Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393: 599-601. doi: 10.1038/31275
|
| [87] |
Hornecker JL, Samollow PB, Robinson ES, et al. (2007) Meiotic sex chromosome inactivation in the marsupial Monodelphis domestica. Genesis 45: 696-708. doi: 10.1002/dvg.20345
|
| [88] |
Kaslow DC, Migeon BR, Persico MG, et al. (1987) Molecular studies of marsupial X chromosomes reveal limited sequence homology of mammalian X-linked genes. Genomics 1: 19-28. doi: 10.1016/0888-7543(87)90100-5
|
| [89] |
Loebel DA, Johnston PG (1996) Methylation analysis of a marsupial X-linked CpG island by bisulfite genomic sequencing. Genome Res 6: 114-123. doi: 10.1101/gr.6.2.114
|
| [90] | Wang X, Douglas KC, Vandeberg JL, et al. (2013) Chromosome-wide profiling of X-chromosome inactivation and epigenetic states in fetal brain and placenta of the opossum, Monodelphis domestica. Genome Res 24: 70-83. |
| [91] |
Loebel DA, Johnston PG (1993) Analysis of DNase 1 sensitivity and methylation of active and inactive X chromosomes of kangaroos (Macropus robustus) by in situ nick translation. Chromosoma 102: 81-87. doi: 10.1007/BF00356024
|
| [92] |
Hellman A, Chess A (2007) Gene body-specific methylation on the active X chromosome. Science 315: 1141-1143. doi: 10.1126/science.1136352
|
| [93] |
Deakin JE, Hore TA, Koina E, et al. (2008) The status of dosage compensation in the multiple X chromosomes of the platypus. PLoS Genet 4: e1000140. doi: 10.1371/journal.pgen.1000140
|
| [94] |
Livernois AM, Waters SA, Deakin JE, et al. (2013) Independent evolution of transcriptional inactivation on sex chromosomes in birds and mammals. PLoS Genet 9: e1003635. doi: 10.1371/journal.pgen.1003635
|
| [95] | Livernois AM (2013) Evolution of transcriptional inactivation on sex chromosomes in birds and mammals: The Australian National University. 145. |
| [96] |
Kondilis-Mangum HD, Wade PA (2013) Epigenetics and the adaptive immune response. Mol Aspects Med 34: 813-825. doi: 10.1016/j.mam.2012.06.008
|
| [97] |
Seita J, Weissman IL (2010) Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2: 640-653. doi: 10.1002/wsbm.86
|
| [98] |
Zediak VP, Wherry EJ, Berger SL (2011) The contribution of epigenetic memory to immunologic memory. Curr Opin Genet Dev 21: 154-159. doi: 10.1016/j.gde.2011.01.016
|
| [99] |
Araki Y, Wang Z, Zang C, et al. (2009) Genome-wide Analysis of Histone Methylation Reveals Chromatin State-Based Regulation of Gene Transcription and Function of Memory CD8(+) T Cells. Immunity 30: 912-925. doi: 10.1016/j.immuni.2009.05.006
|
| [100] |
Barski A, Cuddapah S, Cui K, et al. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129: 823-837. doi: 10.1016/j.cell.2007.05.009
|
| [101] |
Roh TY, Cuddapah S, Cui K, et al. (2006) The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A 103: 15782-15787. doi: 10.1073/pnas.0607617103
|
| [102] |
Roh TY, Cuddapah S, Zhao K (2005) Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 19: 542-552. doi: 10.1101/gad.1272505
|
| [103] |
Roh TY, Ngau WC, Cui K, et al. (2004) High-resolution genome-wide mapping of histone modifications. Nat Biotechnol 22: 1013-1016. doi: 10.1038/nbt990
|
| [104] |
Schones DE, Cui K, Cuddapah S, et al. (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132: 887-898. doi: 10.1016/j.cell.2008.02.022
|
| [105] |
Wang Z, Zang C, Rosenfeld JA, et al. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897-903. doi: 10.1038/ng.154
|
| [106] |
Lim PS, Shannon MF, Hardy K (2010) Epigenetic control of inducible gene expression in the immune system. Epigenomics 2: 775-795. doi: 10.2217/epi.10.55
|
| [107] |
Wei G, Wei L, Zhu J, et al. (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30: 155-167. doi: 10.1016/j.immuni.2008.12.009
|
| [108] |
Lim PS, Hardy K, Bunting KL, et al. (2009) Defining the chromatin signature of inducible genes in T cells. Genome Biol 10: R107. doi: 10.1186/gb-2009-10-10-r107
|
| [109] |
Smith AE, Chronis C, Christodoulakis M, et al. (2009) Epigenetics of human T cells during the G0-->G1 transition. Genome Res 19: 1325-1337. doi: 10.1101/gr.085530.108
|
| [110] |
Barski A, Jothi R, Cuddapah S, et al. (2009) Chromatin poises miRNA- and protein-coding genes for expression. Genome Res 19: 1742-1751. doi: 10.1101/gr.090951.109
|
| [111] |
Belov K, Sanderson CE, Deakin JE, et al. (2007) Characterization of the opossum immune genome provides insights into the evolution of the mammalian immune system. Genome Res 17: 982-991. doi: 10.1101/gr.6121807
|
| [112] |
Deakin JE, Belov K, Curach NC, et al. (2005) High levels of variability in immune response using antigens from two reproductive proteins in brushtail possums. Wildlife Res 32: 1-6. doi: 10.1071/WR03107
|
| [113] | Yadav M (1971) The transmissions of antibodies across the gut of pouch-young marsupials. Immunology 21: 839-851. |
| [114] |
Duncan LG, Nair SV, Deane EM (2009) The marsupial CD8 gene locus: molecular cloning and expression analysis of the alpha and beta sequences in the gray short-tailed opossum (Monodelphis domestica) and the tammar wallaby (Macropus eugenii). Vet Immunol Immunopathol 129: 14-27. doi: 10.1016/j.vetimm.2008.12.003
|
| [115] |
Wong ES, Papenfuss AT, Belov K (2011) Immunome database for marsupials and monotremes. BMC Immunol 12: 48. doi: 10.1186/1471-2172-12-48
|
| [116] |
Duncan L, Webster K, Gupta V, et al. (2010) Molecular characterisation of the CD79a and CD79b subunits of the B cell receptor complex in the gray short-tailed opossum (Monodelphis domestica) and tammar wallaby (Macropus eugenii): Delayed B cell immunocompetence in marsupial neonates. Vet Immunol Immunopathol 136: 235-247. doi: 10.1016/j.vetimm.2010.03.013
|
| [117] |
Duncan LG, Nair SV, Deane EM (2012) Immunohistochemical localization of T-lymphocyte subsets in the developing lymphoid tissues of the tammar wallaby (Macropus eugenii). Dev Comp Immunol 38: 475-486. doi: 10.1016/j.dci.2012.06.015
|
| [118] |
Hussein MF, Badir N, Elridi R, et al. (1978) Effect of Seasonal-Variation on Lymphoid-Tissues of Lizards, Mabuya-Quinquetaeniata Licht and Uromastyx-Aegyptia Forsk. Dev Comp Immunol 2: 469-478. doi: 10.1016/S0145-305X(78)80008-1
|
| [119] |
Hussein MF, Badir N, Elridi R, et al. (1979) Lymphoid-Tissues of the Snake, Spalerosophis-Diadema, in the Different Seasons. Dev Comp Immunol 3: 77-88. doi: 10.1016/S0145-305X(79)80008-7
|
| [120] |
Hussein MF, Badir N, Ridi RE, et al. (1978) Differential Effect of Seasonal-Variation on Lymphoid-Tissue of Lizard, Chalcides-Ocellatus. Dev Comp Immunol 2: 297-309. doi: 10.1016/S0145-305X(78)80072-X
|
| [121] |
Elridi R, Badir N, Elrouby S (1981) Effect of Seasonal-Variations on the Immune-System of the Snake, Psammophis-Schokari. J Exp Zool 216: 357-365. doi: 10.1002/jez.1402160303
|
| [122] |
Hussein MF, Badir N, Elridi R, et al. (1979) Effect of Seasonal-Variation on Immune System of the Lizard, Scincus-Scincus. J Exp Zool 209: 91-96. doi: 10.1002/jez.1402090111
|
| [123] |
Alfoldi J, Di Palma F, Grabherr M, et al. (2011) The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 477: 587-591. doi: 10.1038/nature10390
|
| [124] |
Castoe TA, de Koning AP, Hall KT, et al. (2013) The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A 110: 20645-20650. doi: 10.1073/pnas.1314475110
|
| [125] |
Shaffer HB, Minx P, Warren DE, et al. (2013) The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol 14: R28. doi: 10.1186/gb-2013-14-3-r28
|
| [126] |
Vonk FJ, Casewell NR, Henkel CV, et al. (2013) The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A 110: 20651-20656. doi: 10.1073/pnas.1314702110
|
| [127] |
Wang Z, Pascual-Anaya J, Zadissa A, et al. (2013) The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet 45: 701-706. doi: 10.1038/ng.2615
|
| [128] |
McPherson FJ, Chenoweth PJ (2012) Mammalian sexual dimorphism. Anim Reprod Sci 131: 109-122. doi: 10.1016/j.anireprosci.2012.02.007
|
| [129] |
Kuroki S, Matoba S, Akiyoshi M, et al. (2013) Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science 341: 1106-1109. doi: 10.1126/science.1239864
|
| [130] | Charnier M (1966) [Action of temperature on the sex ratio in the Agama agama (Agamidae, Lacertilia) embryo]. C R Seances Soc Biol Fil 160: 620-622. |
| [131] |
Piferrer F (2013) Epigenetics of sex determination and gonadogenesis. Dev Dyn 242: 360-370. doi: 10.1002/dvdy.23924
|
| [132] |
Gorelick R (2003) Evolution of dioecy and sex chromosomes via methylation driving Muller's ratchet. Biol J Linn Soc 80: 353-368. doi: 10.1046/j.1095-8312.2003.00244.x
|
| [133] |
Eggert C (2004) Sex determination: the amphibian models. Reprod Nutr Dev 44: 539-549. doi: 10.1051/rnd:2004062
|
| [134] |
Martinez-Arguelles DB, Papadopoulos V (2010) Epigenetic regulation of the expression of genes involved in steroid hormone biosynthesis and action. Steroids 75: 467-476. doi: 10.1016/j.steroids.2010.02.004
|
| [135] |
Zhang X, Ho SM (2011) Epigenetics meets endocrinology. J Mol Endocrinol 46: R11-32. doi: 10.1677/JME-10-0053
|
| [136] |
Navarro-Martin L, Vinas J, Ribas L, et al. (2011) DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European sea bass. PLoS Genet 7: e1002447. doi: 10.1371/journal.pgen.1002447
|
| [137] | Ramsey M, Shoemaker C, Crews D (2007) Gonadal expression of Sf1 and aromatase during sex determination in the red-eared slider turtle (Trachemys scripta), a reptile with temperature-dependent sex determination. Differentiation 75: 978-991. |
| [138] |
Matsumoto Y, Buemio A, Chu R, et al. (2013) Epigenetic control of gonadal aromatase (cyp19a1) in temperature-dependent sex determination of red-eared slider turtles. PLoS One 8: e63599. doi: 10.1371/journal.pone.0063599
|
| [139] |
Shao C, Li Q, Chen S, et al. (2014) Epigenetic modification and inheritance in sexual reversal of fish. Genome Res 24: 604-615. doi: 10.1101/gr.162172.113
|
| [140] |
Chen S, Zhang G, Shao C, et al. (2014) Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet 46: 253-260. doi: 10.1038/ng.2890
|
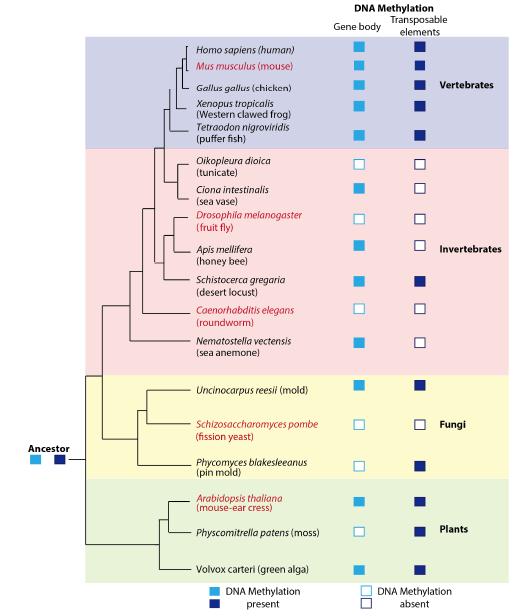
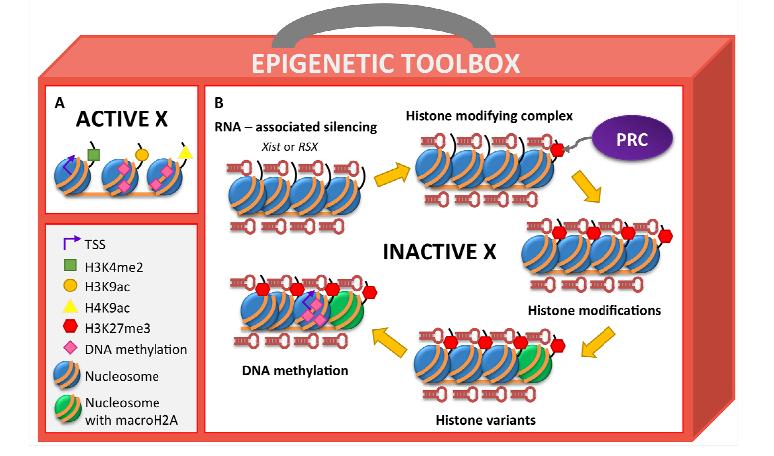
Figures(2) / Tables(2)

Janine E. Deakin, Renae Domaschenz, Pek Siew Lim, Tariq Ezaz, Sudha Rao. Comparative epigenomics: an emerging field with breakthrough potential to understand evolution of epigenetic regulation[J]. AIMS Genetics, 2014, 1(1): 34-54. doi: 10.3934/genet.2014.1.34







 DownLoad:
DownLoad: