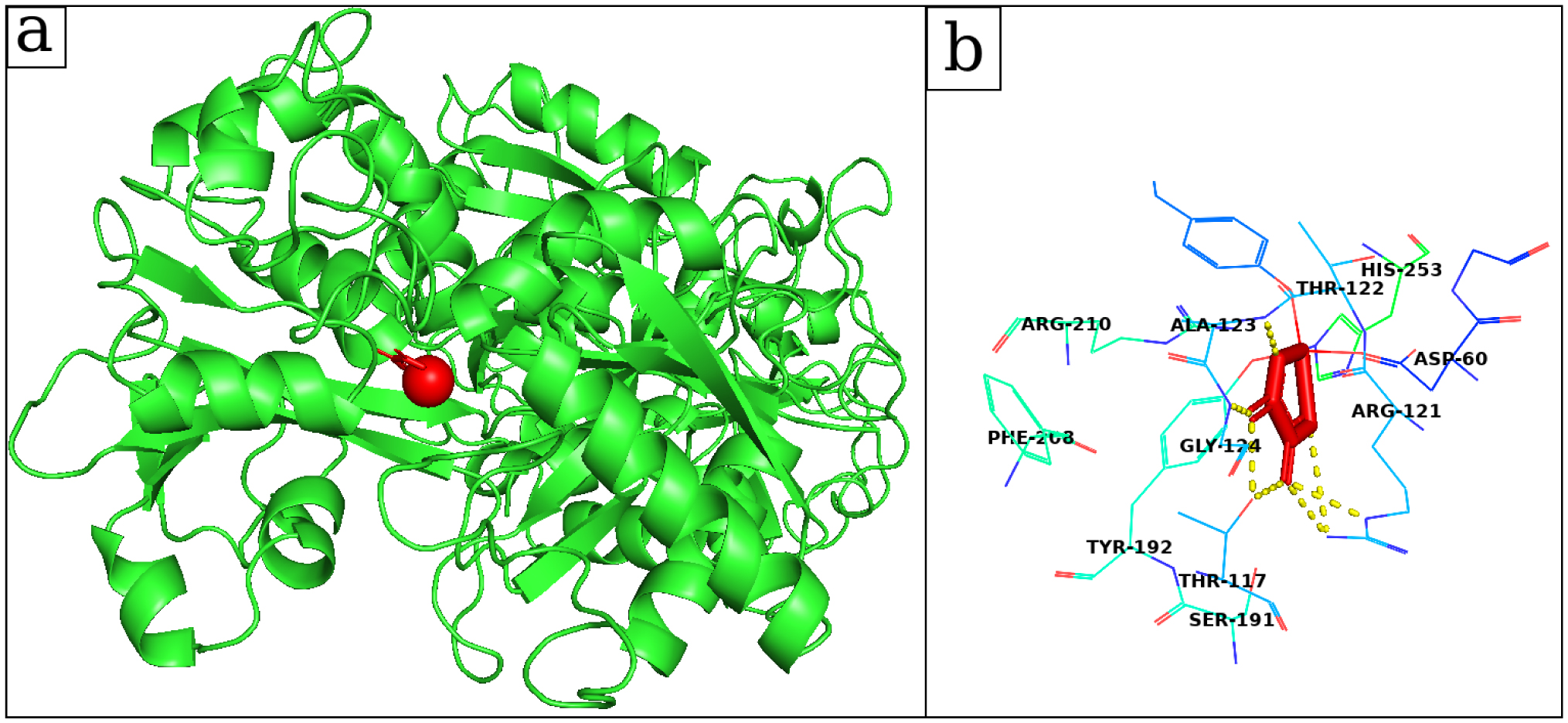
Lactoferrin, a member of the transferrin family, is one of the promoter proteins for calcium oxalate-type kidney stone formation. It exhibits a remarkable ability to interact with metals and oxalate ions. The prevalence of calcium oxalate in kidney stones was confirmed by the Fourier transform infrared spectra. The quantum chemical properties of calcium oxalate and dolichin A calculated by density functional theory and time-dependent density functional theory indicate their potential for hydrogen bonding and nonbonding interactions with the receptor proteins. From molecular docking analysis, the binding free energy of dolichin A was −7.78 kcal/mol, which was the best of twenty-four phytochemicals from Macrotyloma uniflorum, and that of calcium oxalate was −3.86 kcal/mol to lactoferrin. Furthermore, dolichin A having favorable physicochemical and pharmacokinetic properties offers post molecular dynamics molecular mechanics generalized Born surface area free energy of −17.61 ± 4.03 kcal/mol, indicating the strong binding interactions, and, therefore, it acts as a potential inhibitor of the lactoferrin.
Citation: Arjun Acharya, Madan Khanal, Rajesh Maharjan, Kalpana Gyawali, Bhoj Raj Luitel, Rameshwar Adhikari, Deependra Das Mulmi, Tika Ram Lamichhane, Hari Prasad Lamichhane. Quantum chemical calculations on calcium oxalate and dolichin A and their binding efficacy to lactoferrin: An in silico study using DFT, molecular docking, and molecular dynamics simulations[J]. AIMS Biophysics, 2024, 11(2): 142-165. doi: 10.3934/biophy.2024010
Lactoferrin, a member of the transferrin family, is one of the promoter proteins for calcium oxalate-type kidney stone formation. It exhibits a remarkable ability to interact with metals and oxalate ions. The prevalence of calcium oxalate in kidney stones was confirmed by the Fourier transform infrared spectra. The quantum chemical properties of calcium oxalate and dolichin A calculated by density functional theory and time-dependent density functional theory indicate their potential for hydrogen bonding and nonbonding interactions with the receptor proteins. From molecular docking analysis, the binding free energy of dolichin A was −7.78 kcal/mol, which was the best of twenty-four phytochemicals from Macrotyloma uniflorum, and that of calcium oxalate was −3.86 kcal/mol to lactoferrin. Furthermore, dolichin A having favorable physicochemical and pharmacokinetic properties offers post molecular dynamics molecular mechanics generalized Born surface area free energy of −17.61 ± 4.03 kcal/mol, indicating the strong binding interactions, and, therefore, it acts as a potential inhibitor of the lactoferrin.

| [1] | Lameire NH, Bagga A, Cruz D, et al. (2013) Acute kidney injury: an increasing global concern. Lancet 382: 170-179. https://doi.org/10.1016/S0140-6736(13)60647-9 |
| [2] | Ziemba JB, Matlaga BR (2017) Epidemiology and economics of nephrolithiasis. Investig Clin Urol 58: 299-306. https://doi.org/10.4111/icu.2017.58.5.299 |
| [3] | Pak CY, Poindexter JR, Adams-Huet B, et al. (2003) Predictive value of kidney stone composition in the detection of metabolic abnormalities. Am J Med 115: 26-32. https://doi.org/10.1016/S0002-9343(03)00201-8 |
| [4] | Lieske JC, Rule AD, Krambeck AE, et al. (2014) Stone composition as a function of age and sex. Clin J Am Soc Nephrol 9: 2141-2146. https://doi.org/10.2215/cjn.05660614 |
| [5] | Ouyang JM, Zheng H, Deng SP (2006) Simultaneous formation of calcium oxalate (mono-, di-, and trihydrate) induced by potassium tartrate in gelatinous system. J Crys Growth 293: 118-123. https://doi.org/10.1016/j.jcrysgro.2006.05.008 |
| [6] | Rijal R, Sah M, Lamichhane HP, et al. (2022) Quantum chemical calculations of nicotine and caffeine molecule in gas phase and solvent using DFT methods. Heliyon 8: e12494. https://doi.org/10.1016/j.heliyon.2022.e12494 |
| [7] | Aggarwal KP, Narula S, Kakkar M, et al. (2013) Nephrolithiasis: molecular mechanism of renal stone formation and the critical role played by modulators. BioMed Res Int 2013: 292953. https://doi.org/10.1155/2013/292953 |
| [8] | Farmanesh S, Chung J, Sosa RD, et al. (2014) Natural promoters of calcium oxalate monohydrate crystallization. J Am Chem Soc 136: 12648-12657. https://doi.org/10.1021/ja505402r |
| [9] | Åbrink M, Larsson E, Gobl A, et al. (2000) Expression of lactoferrin in the kidney: implications for innate immunity and iron metabolism. Kidney Int 57: 2004-2010. https://doi.org/10.1046/j.1523-1755.2000.00050.x |
| [10] | Baker HM, Anderson BF, Brodie AM, et al. (1996) Anion binding by transferrins: importance of second-shell effects revealed by the crystal structure of oxalate-substituted diferric lactoferrin. Biochemistry 35: 9007-9013. https://doi.org/10.1021/bi960288y |
| [11] | Nageswari P, Swathi K (2023) In silico docking and molecular dynamic (MD) simulations studies of selected phytochemicals against human glycolate oxidase (hGOX) and oxalate oxidase (OxO). Drug Res 73: 459-464. https://doi.org/10.1055/a-2088-3889 |
| [12] | Chattaraj B, Nandi A, Das A, et al. (2023) Inhibitory activity of Enhydra fluctuans lour. on calcium oxalate crystallisation through in silico and in vitro studies. Front Pharmacol 13: 982419. https://doi.org/10.3389/fphar.2022.982419 |
| [13] | Pinipay F, Rokkam R, Botcha S, et al. (2023) Evaluating the anti-urolithiasis potential of Ficus religiosa seed GC MS evaluated phytoconstituents based on their in-vitro antioxidant properties and in-silico ADMET and molecular docking studies. Clin Phytosci 9: 7. https://doi.org/10.1186/s40816-023-00359-2 |
| [14] | Gautam M, Katoch S, Chahota RK (2020) Comprehensive nutritional profiling and activity directed identification of lead antioxidant, antilithiatic agent from Macrotyloma uniflorum (Lam.) Verdc. Food Res Int 137: 109600. https://doi.org/10.1016/j.foodres.2020.109600 |
| [15] | Sharma N, Bisht SS, Gupta S, et al. (2018) Analysis of proteomic diversity and calcium binding protein(s) in seeds of horse gram (Macrotyloma uniflorum) cultivars from Uttarakhand. Int J Pharm Sci Res 9: 3274-3280. http://dx.doi.org/10.13040/IJPSR.0975-8232.9(8).3274-80 |
| [16] | Aditya JP, Bhartiya A, Chahota RK, et al. (2019) Ancient orphan legume horse gram: a potential food and forage crop of future. Planta 250: 891-909. https://doi.org/10.1007/s00425-019-03184-5 |
| [17] | Sousa SF, Fernandes PA, Ramos MJ (2006) Protein–ligand docking: current status and future challenges. Proteins 65: 15-26. https://doi.org/10.1002/prot.21082 |
| [18] | Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10: 449-461. https://doi.org/10.1517/17460441.2015.1032936 |
| [19] | Pan Y, Lu Z, Li C, et al. (2021) Molecular dockings and molecular dynamics simulations reveal the potency of different inhibitors against Xanthine oxidase. ACS Omega 6: 11639-11649. https://doi.org/10.1021/acsomega.1c00968 |
| [20] | Gaussian 16 Revision C.01 Gaussian Inc, Wallingford CT, 2016. Available from: https://gaussian.com/gaussian16/ |
| [21] | GaussView Version 6 Semichem Inc, Shawnee Mission KS, 2019. Available from: https://gaussian.com/gaussview6/ |
| [22] | O'boyle NM, Tenderholt AL, Langner KM (2008) Cclib: a library for package-independent computational chemistry algorithms. J Comput Chem 29: 839-845. https://doi.org/10.1002/jcc.20823 |
| [23] | Jamróz MH (2013) Vibrational energy distribution analysis (VEDA): scopes and limitations. Spectrochim Acta Part A 114: 220-230. https://doi.org/10.1016/j.saa.2013.05.096 |
| [24] | Waterhouse A, Bertoni M, Bienert S, et al. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296-W303. https://doi.org/10.1093/nar/gky427 |
| [25] | Kawsar SM, Mostafa G, Huq E, et al. (2009) Chemical constituents and hemolytic activity of Macrotyloma uniflorum L. Int J Biol Chem 3: 42-48. https://doi.org/10.3923/ijbc.2009.42.48 |
| [26] | Ingle KP, Al-Khayri JM, Chakraborty P, et al. (2020) Bioactive compounds of horse gram (Macrotyloma uniflorum Lam.[Verdc.]). Bioactive Compounds in Underutilized Vegetables and Legumes . Cham: Springer 1-39. https://doi.org/10.1007/978-3-030-44578-2_36-1 |
| [27] | Peter JS, Kappagantu A, Thripureshwari V, et al. (2021) In silico approach to predict the potential binding affinity of the active ingredient of the Macrotyloma uniflorum seed against orphan nuclear receptor. Res J Pharm Technol 14: 694-700. http://dx.doi.org/10.5958/0974-360X.2021.00122.0 |
| [28] | Prasad SK, Singh MK (2015) Horse gram-an underutilized nutraceutical pulse crop: a review. J Food Sci Technol 52: 2489-2499. https://doi.org/10.1007/s13197-014-1312-z |
| [29] | Kim S, Chen J, Cheng T, et al. (2023) PubChem 2023 update. Nucleic Acids Res 51: D1373-D1380. https://doi.org/10.1093/nar/gkac956 |
| [30] | Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Chemical Biology: Methods and Protocols . New York: Humana Press 243-250. https://doi.org/10.1007/978-1-4939-2269-7_19 |
| [31] | Morris GM, Huey R, Lindstrom W, et al. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30: 2785-2791. https://doi.org/10.1002/jcc.21256 |
| [32] | Forli S, Huey R, Pique ME, et al. (2016) Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11: 905-919. https://doi.org/10.1038/nprot.2016.051 |
| [33] | Cosconati S, Forli S, Perryman AL, et al. (2010) Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 5: 597-607. https://doi.org/10.1517/17460441.2010.484460 |
| [34] | Morris GM, Goodsell DS, Halliday RS, et al. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19: 1639-1662. https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14%3C1639::AID-JCC10%3E3.0.CO;2-B |
| [35] | Huey R, Morris GM, Forli S (2012) Using AutoDock 4 and AutoDock vina with AutoDockTools: a tutorial. The Scripps Research Institute Molecular Graphics Laboratory 10550: 92037. |
| [36] | Lamichhane TR, Ghimire MP (2021) Evaluation of SARS-CoV-2 main protease and inhibitor interactions using dihedral angle distributions and radial distribution function. Heliyon 7: e08220. https://doi.org/10.1016/j.heliyon.2021.e08220 |
| [37] | Bell EW, Zhang Y (2019) DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J Cheminform 11: 1-9. https://doi.org/10.1186/s13321-019-0362-7 |
| [38] | The PyMOL Molecular Graphics System, Version 1.8, 2015. Available from: https://pymol.org/ |
| [39] | Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 51: 2778-2786. https://doi.org/10.1021/ci200227u |
| [40] | Visualizer, Biovia Discovery Studio, Dassault Systemes; San Diego, CA, USA Version v21.1.0.20298, 2021. Available from: https://www.3ds.com/products/biovia/discovery-studio |
| [41] | Abraham MJ, Murtola T, Schulz R, et al. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1: 19-25. https://doi.org/10.1016/j.softx.2015.06.001 |
| [42] | Zoete V, Cuendet MA, Grosdidier A, et al. (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32: 2359-2368. https://doi.org/10.1002/jcc.21816 |
| [43] | Brooks BR, Brooks CL, Mackerell AD, et al. (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30: 1545-1614. https://doi.org/10.1002/jcc.21287 |
| [44] | Jorgensen WL, Chandrasekhar J, Madura JD, et al. (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926-935. https://doi.org/10.1063/1.445869 |
| [45] | Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7: 42717. https://doi.org/10.1038/srep42717 |
| [46] | Yang H, Lou C, Sun L, et al. (2019) admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35: 1067-1069. https://doi.org/10.1093/bioinformatics/bty707 |
| [47] | Banerjee P, Eckert AO, Schrey AK, et al. (2018) ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 46: W257-W263. https://doi.org/10.1093/nar/gky318 |
| [48] | Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, et al. (2021) gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput 17: 6281-6291. https://doi.org/10.1021/acs.jctc.1c00645 |
| [49] | Lien EJ, Guo ZR, Li RL, et al. (1982) Use of dipole moment as a parameter in drug–receptor interaction and quantitative structure–activity relationship studies. J Pharm Sci 71: 641-655. https://doi.org/10.1002/jps.2600710611 |
| [50] | Barim E, Akman F (2019) Synthesis, characterization and spectroscopic investigation of N-(2-acetylbenzofuran-3-yl) acrylamide monomer: Molecular structure, HOMO–LUMO study, TD-DFT and MEP analysis. J Mol Struct 1195: 506-513. https://doi.org/10.1016/j.molstruc.2019.06.015 |
| [51] | Mishra R, Joshi BD, Srivastava A, et al. (2014) Quantum chemical and experimental studies on the structure and vibrational spectra of an alkaloid–Corlumine. Spectrochim Acta A 118: 470-480. https://doi.org/10.1016/j.saa.2013.09.015 |
| [52] | Fouad R, Adly OM (2021) Novel Cu2+ and Zn2+ nanocomplexes drug based on hydrazone ligand bearings chromone and triazine moieties: structural, spectral, DFT, molecular docking and cytotoxic studies. J Mol Struct 1225: 129158. https://doi.org/10.1016/j.molstruc.2020.129158 |
| [53] | Pearson RG (1988) Absolute electronegativity and hardness: application to inorganic chemistry. Inorg Chem 27: 734-740. https://doi.org/10.1021/ic00277a030 |
| [54] | Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103: 1793-1874. https://doi.org/10.1021/cr990029p |
| [55] | Padmanabhan J, Parthasarathi R, Subramanian V, et al. (2007) Electrophilicity-based charge transfer descriptor. J Phys Chem A 111: 1358-1361. https://doi.org/10.1021/jp0649549 |
| [56] | Parr RG, Szentpály L, Liu S (1999) Electrophilicity index. J Am Chem Soc 121: 1922-1924. https://doi.org/10.1021/ja983494x |
| [57] | Demircioğlu Z, Kaştaş ÇA, Büyükgüngör O (2015) Theoretical analysis (NBO, NPA, Mulliken population method) and molecular orbital studies (hardness, chemical potential, electrophilicity and Fukui function analysis) of (E)-2-((4-hydroxy-2-methylphenylimino) methyl)-3-methoxyphenol. J Mol Struct 1091: 183-195. https://doi.org/10.1016/j.molstruc.2015.02.076 |
| [58] | Pople JA, Schlegel HB, Krishnan R, et al. (1981) Molecular orbital studies of vibrational frequencies. Int J Quantum Chem 20: 269-278. https://doi.org/10.1002/qua.560200829 |
| [59] | Yoshida H, Ehara A, Matsuura H (2000) Density functional vibrational analysis using wavenumber-linear scale factors. Chem Phys Lett 325: 477-483. https://doi.org/10.1016/S0009-2614(00)00680-1 |
| [60] | Abel R, Young T, Farid R, et al. (2008) Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc 130: 2817-2831. https://doi.org/10.1021/ja0771033 |
| [61] | AlRabiah H, Muthu S, Al-Omary F, et al. (2017) Molecular structure, vibrational spectra, NBO, Fukui function, HOMO-LUMO analysis and molecular docking study of 6-[(2-methylphenyl) sulfanyl]-5-propylpyrimidine-2, 4 (1H, 3H)-dione. Maced J Chem Chem Eng 36: 59-80. https://doi.org/10.20450/mjcce.2017.1001 |
| [62] | Bitew M, Desalegn T, Demissie TB, et al. (2021) Pharmacokinetics and drug-likeness of antidiabetic flavonoids: molecular docking and DFT study. PLoS One 16: e0260853. https://doi.org/10.1371/journal.pone.0260853 |
| [63] | Lipinski CA, Lombardo F, Dominy BW, et al. (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 64: 4-17. https://doi.org/10.1016/j.addr.2012.09.019 |
 biophy-11-02-010-s001.pdf biophy-11-02-010-s001.pdf |
 |












Figures(13) / Tables(8)

Arjun Acharya, Madan Khanal, Rajesh Maharjan, Kalpana Gyawali, Bhoj Raj Luitel, Rameshwar Adhikari, Deependra Das Mulmi, Tika Ram Lamichhane, Hari Prasad Lamichhane. Quantum chemical calculations on calcium oxalate and dolichin A and their binding efficacy to lactoferrin: An in silico study using DFT, molecular docking, and molecular dynamics simulations[J]. AIMS Biophysics, 2024, 11(2): 142-165. doi: 10.3934/biophy.2024010







 DownLoad:
DownLoad: