Citation: Mehdi Khoobdel, Davoud Keshavarzi, Seyed Hassan Mossa-Kazemi, Hossein Sobati. Species diversity of mosquitoes of the Genus Culex (Diptera, Culicidae) in the coastal areas of the Persian Gulf[J]. AIMS Public Health, 2019, 6(2): 99-106. doi: 10.3934/publichealth.2019.2.99

| [1] |
Kramer LD, Ebel GD (2003) Dynamics of flavivirus infection in mosquitoes. Adv Virus Res 60: 187–232. doi: 10.1016/S0065-3527(03)60006-0

|
| [2] |
Keshavarzi D, Soltani Z, Ebrahimi M, et al. (2017) Monthly prevalence and diversity of mosquitoes (Diptera; Culicidae) in Fars Province, Southern Iran. Asian Pac J Trop Dis 7: 112–120. doi: 10.12980/apjtd.7.2017D6-369
|
| [3] | Azari‐Hamidian S, Yaghoobi‐Ershadi M, Javadian E, et al. (2009) Distribution and ecology of mosquitoes in a focus of dirofilariasis in northwestern Iran, with the first finding of filarial larvae in naturally infected local mosquitoes. Med Vet Entomol 23: 111–121. |
| [4] |
Diallo D, Sall AA, Diagne CT, et al. (2014) Zika virus emergence in mosquitoes in southeastern Senegal. PloS one 9: e109442. doi: 10.1371/journal.pone.0109442
|
| [5] | Chinikar S, Ghiasi SM, Moradi A, et al. (2010) Laboratory Detection Facility of Dengue Fever (DF) in Iran: The First Imported Case. Int J Infect Dis 8: 1–2. |
| [6] | Khoobdel M, Vatandoost H, Abaei MR (2008) Arthropod borne diseases in imposed war during 1980–88. Irannian J Arthropod-Born Dis 2: 24–32. |
| [7] |
Fakour S, Naserabadi S, Ahmadi E (2017) The first positive serological study on rift valley fever in ruminants of Iran. J Vector Borne Dis 54: 348–352. doi: 10.4103/0972-9062.225840
|
| [8] | Khoobdel M, JonaidiJafari N, Izadi M (2016) Is the Zica threatening the Iran and others Middle East countries?. Iranian J Mil Med 17: 187–190. [Persian]. |
| [9] | Khoobdel M, Azari-Hamidian S, Hanafi-Bojd AA (2012) Mosquito fauna (Diptera: Culicidae) of the Iranian islands in the Persian Gulf II. Greater Tonb, Lesser Tonb and Kish Islands. J Nat Hist 46: 1939–1945. |
| [10] |
Jafari S, Khaksar Z (1996) Prevalence of Dirofilaria immitis in dogs of Fars province of Iran. J Appl Anim Res 9: 27–31. doi: 10.1080/09712119.1996.9706101
|
| [11] |
Ahmadnejad F, Otarod V, Fallah M, et al. (2011) Spread of West Nile virus in Iran: a cross-sectional serosurvey in equines, 2008–2009. Epidemiol Infect 139: 1587–1593. doi: 10.1017/S0950268811000173
|
| [12] |
Hamer GL, Kitron UD, Brawn JD, et al. (2008) Culex pipiens (Diptera: Culicidae): a bridge vector of West Nile virus to humans. J Med Entomol 45: 125–128. doi: 10.1093/jmedent/45.1.125
|
| [13] | Guedes DR, Paiva MH, Donato MM, et al. (2017) Zika virus replication in the mosquito Culex quinquefasciatus in Brazil. Emerg Microbes Infect 6: e69. |
| [14] | Azari-Hamidian S, Harbach RE (2009) Keys to the adult females and fourth-instar larvae of the mosquitoes of Iran (Diptera: Culicidae). Zootaxa 2078: 1–33. |
| [15] | Magurran AE (1988) Why diversity? In: Ecological diversity and its measurement. 1 Ed., Dordrecht: Springer, 1–5. |
| [16] |
Zakhia R, Mousson L, Vazeille M, et al. (2018) Experimental transmission of West Nile Virus and Rift Valley Fever Virus by Culex pipiens from Lebanon. PLOS Negl Trop Dis 12: e0005983. doi: 10.1371/journal.pntd.0005983
|
| [17] | Soltani Z, Keshavarzi D, Ebrahimi M, et al. (2017) The fauna and active season of mosquitoes in west of Fars Province, Southwest of Iran. Arch Razi Inst 72: 203–238. |
| [18] |
Bravo‐Barriga D, Gomes B, Almeida AP, et al. (2017) The mosquito fauna of the western region of Spain with emphasis on ecological factors and the characterization of Culex pipiens forms. J Vector Ecol 42: 136–147. doi: 10.1111/jvec.12248
|
| [19] | Ferreira-de-Freitas V, França RM, Bartholomay LC, et al. (2017) Contribution to the Biodiversity Assessment of Mosquitoes (Diptera: Culicidae) in the Atlantic Forest in Santa Catarina, Brazil. J Med Entomol 54: 368–376. |
| [20] | MacDonald G (2002) Biogeography: introduction to space, time and life, 1 Ed., New York: John Wiley and Sons, 420–428. |
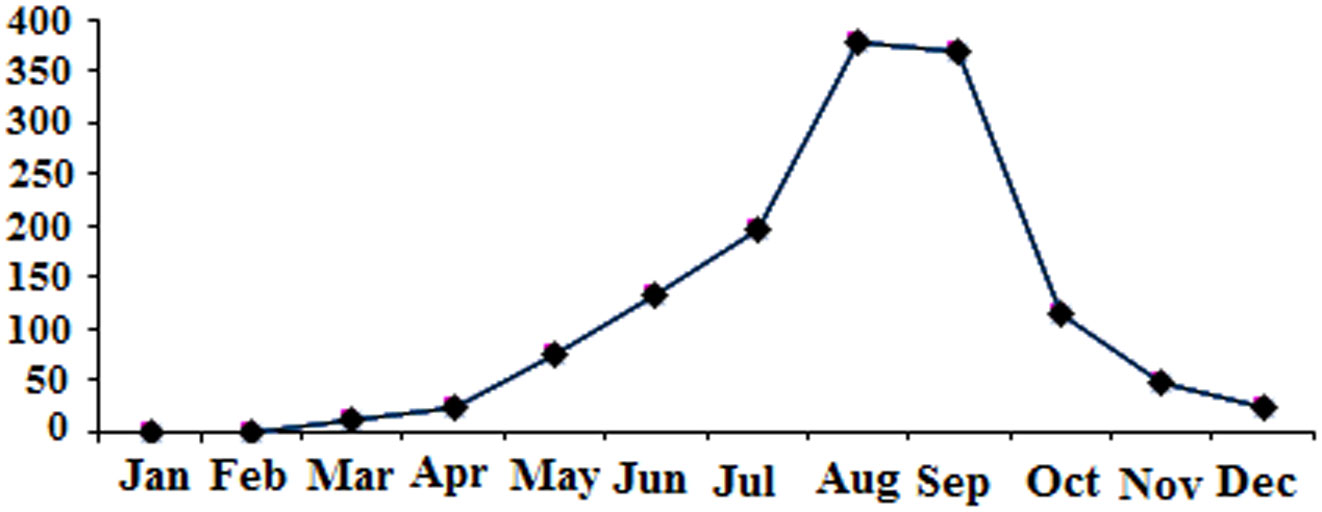
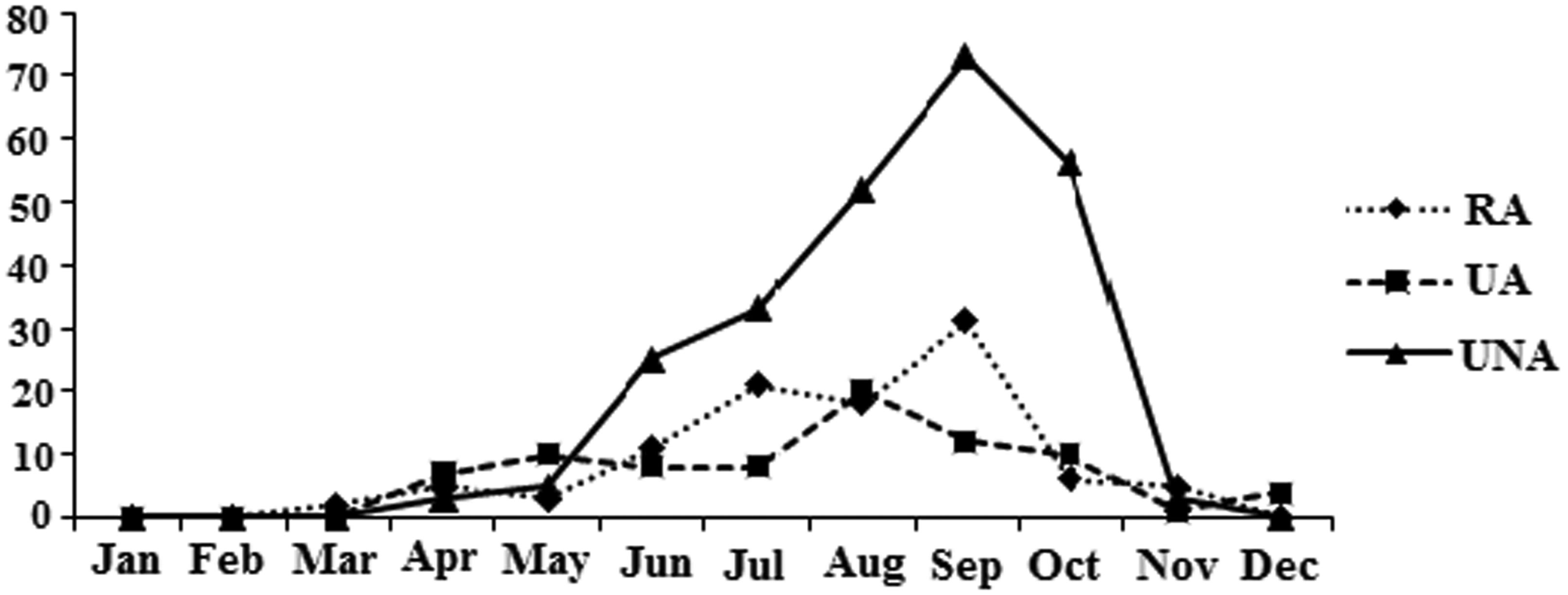
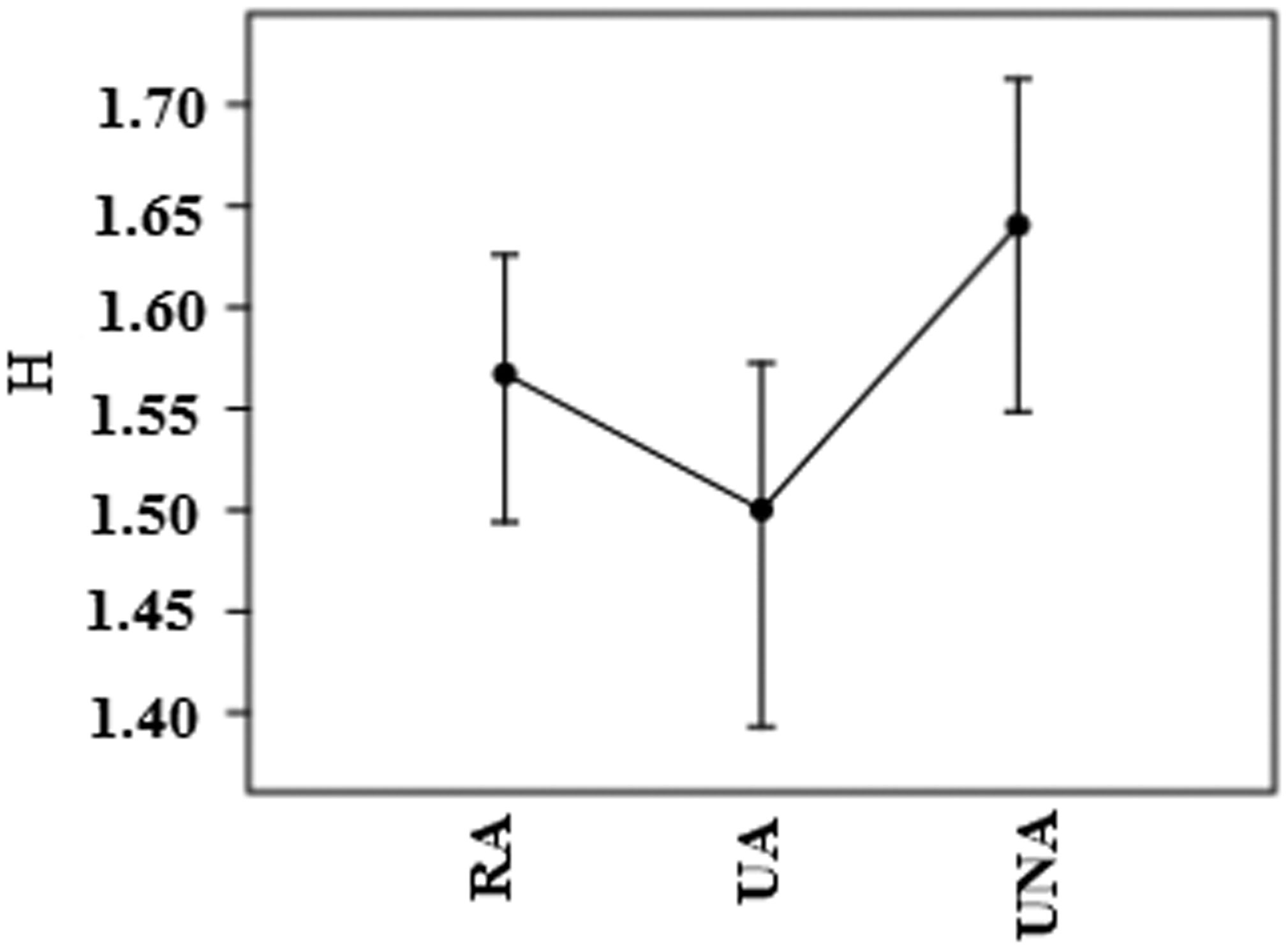
Figures(3) / Tables(2)

Mehdi Khoobdel, Davoud Keshavarzi, Seyed Hassan Mossa-Kazemi, Hossein Sobati. Species diversity of mosquitoes of the Genus Culex (Diptera, Culicidae) in the coastal areas of the Persian Gulf[J]. AIMS Public Health, 2019, 6(2): 99-106. doi: 10.3934/publichealth.2019.2.99







 DownLoad:
DownLoad: