Citation: Hafizah Abdul Halim Yun, Valerie Dupont. Thermodynamic analysis of methanation of palm empty fruit bunch (PEFB) pyrolysis oil with and without in situ CO2 sorption[J]. AIMS Energy, 2015, 3(4): 774-797. doi: 10.3934/energy.2015.4.774

| [1] |
Balat M (2008) Mechanisms of thermochemical biomass conversion processes. Part 1: Reactions of pyrolysis. Energ source part a 30: 620-635. doi: 10.1080/15567030600817258

|
| [2] |
Sulaiman F, Abdullah N (2011) Optimum conditions for maximizing pyrolysis liquids of oil palm empty fruit bunches. Energy 36: 2352-2359. doi: 10.1016/j.energy.2010.12.067
|
| [3] |
Mohan D, Pittman CU, Steele PH (2006) Pyrolysis of wood/biomass for bio-oil: A critical review. Energ fuel 20: 848-889. doi: 10.1021/ef0502397
|
| [4] |
Bridgewater AV (2012) Review of fast pyrolysis of biomass and product upgrading. Biomass bioenerg 38: 68-94. doi: 10.1016/j.biombioe.2011.01.048
|
| [5] |
Zhang R, Cummer K, Suby A, et al. (2005) Biomass-derived hydrogen from an air-blown gasifier. Fuel process technol 86: 861-874. doi: 10.1016/j.fuproc.2004.09.001
|
| [6] | Sulaiman F, Abdullah N, Gerhauser H, et al. (2011) Review: An outlook of Malaysian energy, oil palm industry and its utilization of wastes as useful resources. Biomass bioenerg 35: 3775-3786. |
| [7] | Badger PC, Fransham P (2006) Use of mobile fast pyrolysis plants to densify biomass and reduce biomass handling costs-a preliminary assessment. Biomass bioenerg 20: 321-325. |
| [8] |
Czernik S, Bridgewater AV (2004) Overview of applications of biomass fast pyrolysis oil. Energ fuel 18: 590-598. doi: 10.1021/ef034067u
|
| [9] |
Kopyscinski J, Schildhauer TJ, Biollaz SMA (2010) Production of synthetic natural gas (SNG) from coal and dry biomass: A technology review from 1950 to 2009. Fuel 89: 1763-1783. doi: 10.1016/j.fuel.2010.01.027
|
| [10] | National Renewable Energy Laboratory (US). Energy analysis biogas potential in the United States. Colorado (US): U.S. Department of Energy (US); 2013. 4 p. (NREL publication; no. NREL/FS-6A20-60178). |
| [11] | AEBIOM European Biomass Association. A biogas road map for Europe. Belgium: Renewable Energy House, Rue d’Arlon, Brussels; 2009 October. Available from: http://www.aebiom.org/IMG/pdf/Brochure_BiogasRoadmap_WEB.pdf. |
| [12] | Kelleher Environmental. Benefits to the economy, environment and energy. Canada: The Biogas Association; 2013 November. Available from: http://www.biogasassociation.ca/bioExp/images/uploads/documents/2013/resources/Canadian_Biogas_Study_Summary.pdf. |
| [13] | BP. BP Statistical Review of World Energy June 2015. UK: Heriot-Watt University; 2015 June. Available from: http://www.bp.com/content/dam/bp-country/de_de/PDFs/brochures/bp-statistical-review-of-world-energy-2015-full-report.pdf |
| [14] | Suruhanjaya Tenaga (Energy Commission). National Energy Balance 2013. Malaysia: Energy Commission, Putrajaya; 2014 March. Available from: http://meih.st.gov.my/documents/10620/167a0433-510c-4a4e-81cd-fb178dcb156f |
| [15] | Beer T, Grant T, Brown R. Life-cycle emissions analysis of alternative fuels for heavy vehicles. Australia: Australian Greenhouse Office; 2000. 148p. (CSIRO Atmospheric Research report; no. C/0411/1.1/F2). |
| [16] | Van der Meijden CM, Veringa H, Vreugdenhill BJ, et al. (2009) Production of bio-methane from woody biomass. Available from: http://www.ecn.nl/docs/library/report/2009/m09086.pdf. |
| [17] |
Xie H, Yu Q, Wei M, et al. (2015) Hydrogen production from steam reforming of simulated bio-oil over Ce-Ni/Co catalyst with in continuous CO2 capture. Int j hydrogen energ 40: 1420-1428. doi: 10.1016/j.ijhydene.2014.11.137
|
| [18] |
Md Zin R, Lea-Langton A, Dupont V, et al. (2012) High hydrogen yield and purity from palm empty fruit bunch and pine pyrolysis oils. Int j hydrogen energ 37: 10627-10638. doi: 10.1016/j.ijhydene.2012.04.064
|
| [19] | Remon J, Broust F, Volle G, et al. (2015) Hydrogen production from pine and poplar bio-oils by catalytic steam reforming. Influence of the bio-oil composition on the process. Int j hydrogen energ 40: 5593-5608. |
| [20] |
Xie H, Yu Q, Wang K, et al. (2014) Thermodynamic analysis of hydrogen production from model compounds of bio-oil through steam reforming. Environ prog sustain energy 33: 1008-1016. doi: 10.1002/ep.11846
|
| [21] | Department of Energy and Climate Change. Digest of United Kingdom Energy Statistics 2014. London: The National Archives; 2014. Available from: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/338750/DUKES_2014_printed.pdf |
| [22] |
Oh TH, Pang SH, Chua SC (2010) Energy policy and alternative energy in Malaysia: Issues and challenges for sustainable growth. Renew sust energ rev 14: 1241-1252. doi: 10.1016/j.rser.2009.12.003
|
| [23] |
Hosseini SE, Abdul Wahid M (2013) Feasibility study of biogas production and utilization as a source of renewable energy in Malaysia. Renew sust energ rev 19: 454-462. doi: 10.1016/j.rser.2012.11.008
|
| [24] |
Subramaniam V, Ngan MA, May CY, et al. (2008) Environmental performance of the milling process of Malaysian palm oil using the life cycle assessment approach. American j environ sci 4: 310-315. doi: 10.3844/ajessp.2008.310.315
|
| [25] |
Prasertsan S, Prasertsan P (1996) Biomass residues from palm oil mills in Thailand: An overview on quantity and potential usage. Biomass bioenerg 11: 387-395. doi: 10.1016/S0961-9534(96)00034-7
|
| [26] | Van der Meijden CM, Veringa HJ, Rabou LPLM (2010) The production of synthetic natural gas (SNG): A comparison of three wood gasification systems for energy balance and overall efficiency. Biomass bioenerg 32: 302-311 |
| [27] |
Ni M, Leung DYC, Leung MKH, et al. (2006) An overview of hydrogen production from biomass. Fuel process technol 87: 461-472. doi: 10.1016/j.fuproc.2005.11.003
|
| [28] |
Bridgewater AV (2012) Review of fast pyrolysis of biomass and product upgrading. Biomass bioenerg 38: 68-94. doi: 10.1016/j.biombioe.2011.01.048
|
| [29] |
Sukiran MA, Chin CM, Abu Bakar NK (2009) Bio-oils from pyrolysis of oil palm empty fruit bunches. American j appl sci 6: 869-875. doi: 10.3844/ajassp.2009.869.875
|
| [30] | Abdullah N, Sulaiman F, Gerhauser H (2011) Characterisation of oil palm empty fruit bunches for fuel application. J phys sci 22: 1-24. |
| [31] | Abdullah N, Gerhauser H, Bridgwater AV (2007) Bio-oil from fast pyrolysis of oil palm empty fruit bunches. J phys sci 18: 57-74. |
| [32] |
Gu H, Song G, Xiao J, et al. (2013) Thermodynamic analysis of the biomass-to-synthetic natural gas using chemical looping technology with CaO sorbent. Energ fuel 27: 4695-4704. doi: 10.1021/ef4007593
|
| [33] | Zeleznik FJ, Gordon S. An analytical investigation of three general methods of calculating chemical-equilibrium compositions. Ohio (US): National Aeronautics and Space Administration (US); 1960. 37p. (NASA publication; no. NASA TN D-43). |
| [34] | Gordon S, McBride BJ. Computer program for calculation of complex chemical equilibrium compositions and applications: I. Analysis. Ohio (US): National Aeronautics and Space Administration (US); 1994. 58p. (NASA publication; no. NASA RP-1311). |
| [35] |
Dupont V, Twigg MV, Rollinson AN, et al. (2013) Thermodynamics of hydrogen production from urea by steam reforming with and without in situ carbon dioxide sorption. Int j hydrogen energ 38: 10260-10269. doi: 10.1016/j.ijhydene.2013.06.062
|
| [36] |
Wang Q, Luo J, Zhong Z, et al. (2011) CO2 capture by solid adsorbents and their applications: current status and new trends. Energ environ sci 4: 42-55. doi: 10.1039/C0EE00064G
|
| [37] |
Blamey J, Manovic V, Anthony EJ, et al. (2015) On steam hydration of CaO-based sorbent cycled for CO2 capture. Fuel 150: 269-277. doi: 10.1016/j.fuel.2015.02.026
|
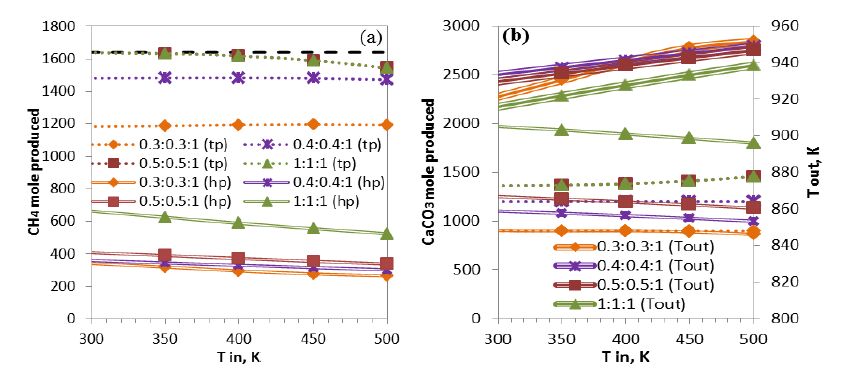
Figures(9) / Tables(7)

Hafizah Abdul Halim Yun, Valerie Dupont. Thermodynamic analysis of methanation of palm empty fruit bunch (PEFB) pyrolysis oil with and without in situ CO2 sorption[J]. AIMS Energy, 2015, 3(4): 774-797. doi: 10.3934/energy.2015.4.774







 DownLoad:
DownLoad: