To identify the characteristics and subtypes of depressive symptoms and explore the relationship between depressive subtypes and age among Chinese female breast cancer patients.
In this cross-sectional study, 566 breast cancer patients were recruited from three tertiary comprehensive hospital in Shandong Province, China through convenient sampling from April 2013 to June 2019. Depressive symptoms were measured using the Patient Health Questionnaire-9 (PHQ-9). Data analyses included descriptive analyses, latent class analysis.
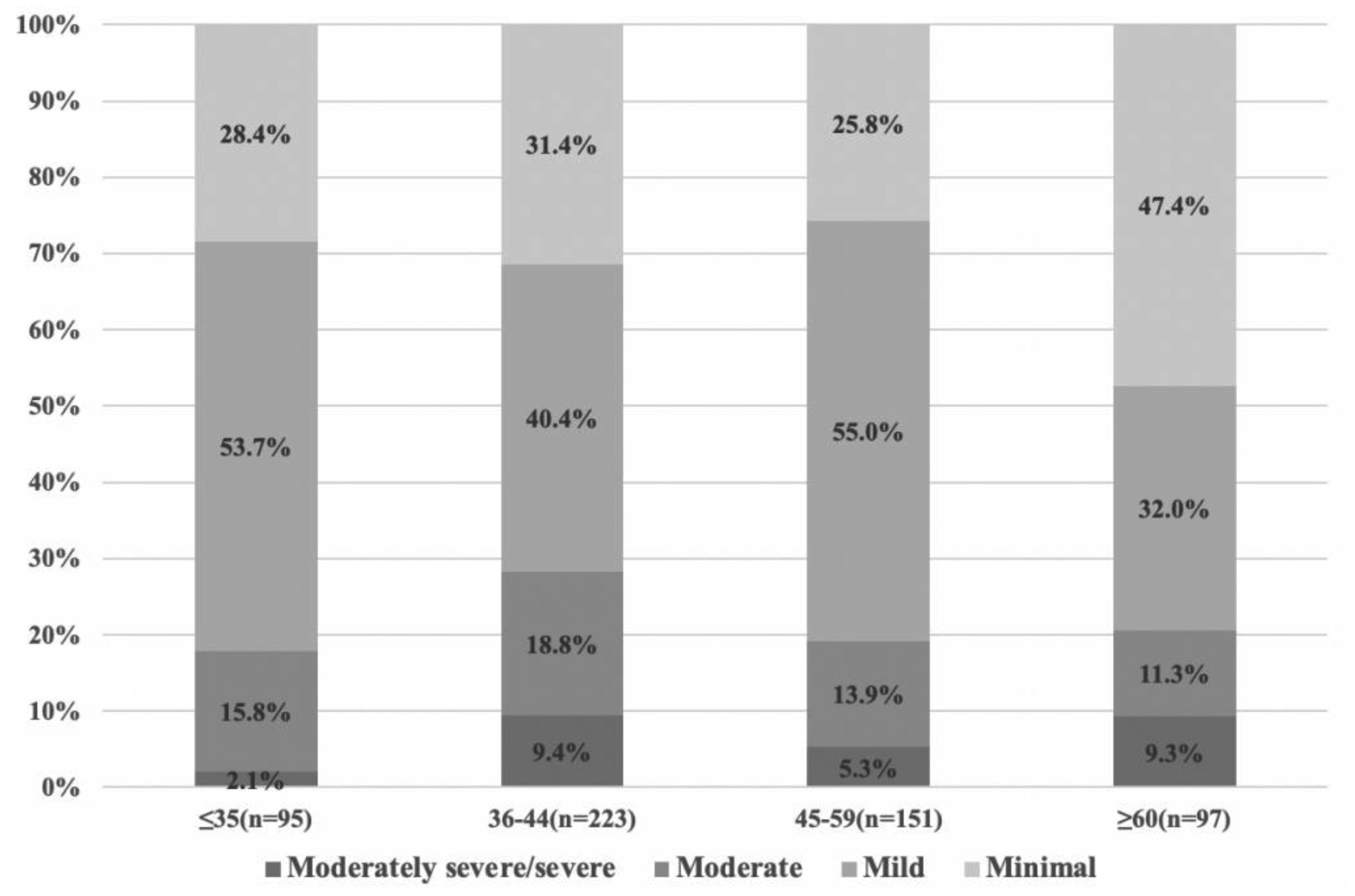
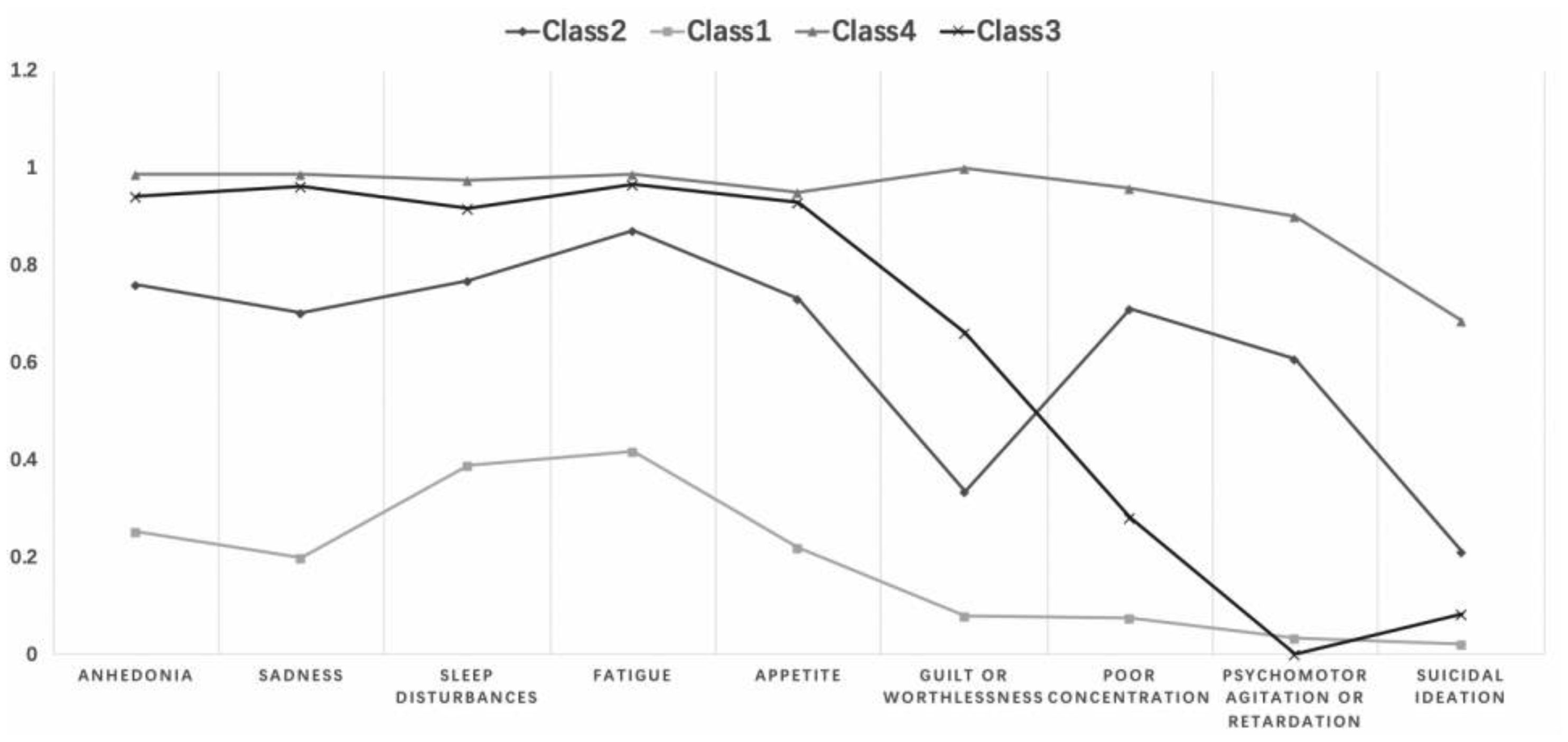
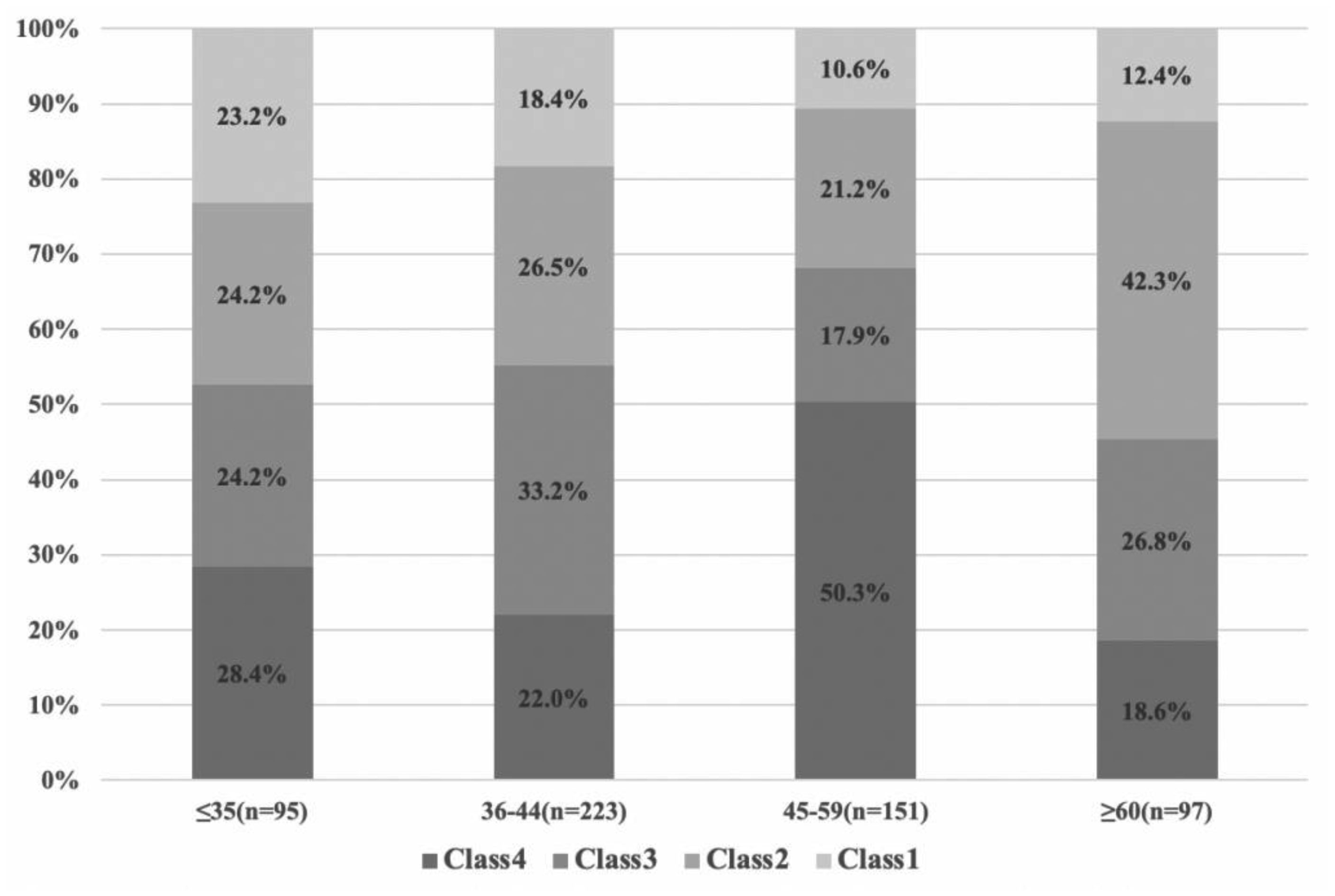
There were significant differences in specific depressive symptoms by age group, but no significant difference in total scores on PHQ-9. The depressive subtypes were severe (Class 4), relatively severe (Class 3; with lower psychomotor agitation/retardation and suicidal ideation), moderate (Class 2; with higher psychomotor agitation/retardation and suicidal ideation), and mild depressive symptoms (Class 1). The distribution of depression subtypes is different in various age groups. In the 45–59 age groups, severe symptoms subtype showed the highest ratios (i.e. 50.3%).
This is the first study that analyses depressive symptom characteristics and identifies depressive subtypes in Chinese women with breast cancer across ages to explore symptom heterogeneity. Our findings can contribute to identifying the mechanisms behind these relationships and developing targeted interventions for patients with specific depressive subtypes.
Citation: Yanyan Li, Hong Liu, Yaoyao Sun, Jie Li, Yanhong Chen, Xuan Zhang, Juan Wang, Liuliu Wu, Di Shao, Fenglin Cao. Characteristics and subtypes of depressive symptoms in Chinese female breast cancer patients of different ages: a cross-sectional study[J]. AIMS Public Health, 2021, 8(4): 691-703. doi: 10.3934/publichealth.2021055
To identify the characteristics and subtypes of depressive symptoms and explore the relationship between depressive subtypes and age among Chinese female breast cancer patients.
In this cross-sectional study, 566 breast cancer patients were recruited from three tertiary comprehensive hospital in Shandong Province, China through convenient sampling from April 2013 to June 2019. Depressive symptoms were measured using the Patient Health Questionnaire-9 (PHQ-9). Data analyses included descriptive analyses, latent class analysis.
There were significant differences in specific depressive symptoms by age group, but no significant difference in total scores on PHQ-9. The depressive subtypes were severe (Class 4), relatively severe (Class 3; with lower psychomotor agitation/retardation and suicidal ideation), moderate (Class 2; with higher psychomotor agitation/retardation and suicidal ideation), and mild depressive symptoms (Class 1). The distribution of depression subtypes is different in various age groups. In the 45–59 age groups, severe symptoms subtype showed the highest ratios (i.e. 50.3%).
This is the first study that analyses depressive symptom characteristics and identifies depressive subtypes in Chinese women with breast cancer across ages to explore symptom heterogeneity. Our findings can contribute to identifying the mechanisms behind these relationships and developing targeted interventions for patients with specific depressive subtypes.

| [1] |
Yap YS, Lu YS, Tamura K, et al. (2019) Insights Into Breast Cancer in the East vs the West: A Review. JAMA Oncol 5: 1489-1496. doi: 10.1001/jamaoncol.2019.0620

|
| [2] |
Chen W, Zheng R, Baade PD, et al. (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66: 115-132. doi: 10.3322/caac.21338
|
| [3] |
Zeng H, Chen W, Zheng R, et al. (2018) Changing cancer survival in China during 2003–15: a pooled analysis of 17 population-based cancer registries. Lancet Glob Health 6: e555-e567. doi: 10.1016/S2214-109X(18)30127-X
|
| [4] |
Gold SM, Kohler-Forsberg O, Moss-Morris R, et al. (2020) Comorbid depression in medical diseases. Nat Rev Dis Primers 6: 69. doi: 10.1038/s41572-020-0200-2
|
| [5] |
Krebber AM, Buffart LM, Kleijn G, et al. (2014) Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology 23: 121-130. doi: 10.1002/pon.3409
|
| [6] | Pratt LA, Brody DJ (2014) Depression in the U.S. household population, 2009–2012. NCHS Data Brief 1-8. |
| [7] |
Pilevarzadeh M, Amirshahi M, Afsargharehbagh R, et al. (2019) Global prevalence of depression among breast cancer patients: a systematic review and meta-analysis. Breast Cancer Res Treat 176: 519-533. doi: 10.1007/s10549-019-05271-3
|
| [8] |
Walker J, Hansen CH, Martin P, et al. (2014) Prevalence, associations, and adequacy of treatment of major depression in patients with cancer: a cross-sectional analysis of routinely collected clinical data. Lancet Psychiatry 1: 343-350. doi: 10.1016/S2215-0366(14)70313-X
|
| [9] |
Mausbach BT, Schwab RB, Irwin SA (2015) Depression as a predictor of adherence to adjuvant endocrine therapy (AET) in women with breast cancer: a systematic review and meta-analysis. Breast Cancer Res Treat 152: 239-246. doi: 10.1007/s10549-015-3471-7
|
| [10] |
Markovitz LC, Drysdale NJ, Bettencourt BA (2017) The relationship between risk factors and medication adherence among breast cancer survivors: What explanatory role might depression play? Psychooncology 26: 2294-2299. doi: 10.1002/pon.4362
|
| [11] |
Chan CM, Wan Ahmad WA, Yusof MM, et al. (2015) Effects of depression and anxiety on mortality in a mixed cancer group: a longitudinal approach using standardised diagnostic interviews. Psychooncology 24: 718-725. doi: 10.1002/pon.3714
|
| [12] |
Grotmol KS, Lie HC, Hjermstad MJ, et al. (2017) Depression-A Major Contributor to Poor Quality of Life in Patients With Advanced Cancer. J Pain Symptom Manage 54: 889-897. doi: 10.1016/j.jpainsymman.2017.04.010
|
| [13] |
Hasson-Ohayon I, Goldzweig G, Dorfman C, et al. (2014) Hope and social support utilisation among different age groups of women with breast cancer and their spouses. Psychol Health 29: 1303-1319. doi: 10.1080/08870446.2014.929686
|
| [14] |
Naik H, Leung B, Laskin J, et al. (2020) Emotional distress and psychosocial needs in patients with breast cancer in British Columbia: younger versus older adults. Breast Cancer Res Treat 179: 471-477. doi: 10.1007/s10549-019-05468-6
|
| [15] |
Avis NE, Levine B, Naughton MJ, et al. (2012) Explaining age-related differences in depression following breast cancer diagnosis and treatment. Breast Cancer Res Treat 136: 581-591. doi: 10.1007/s10549-012-2277-0
|
| [16] |
Vazquez D, Rosenberg S, Gelber S, et al. (2020) Posttraumatic stress in breast cancer survivors diagnosed at a young age. Psychooncology 29: 1312-1320. doi: 10.1002/pon.5438
|
| [17] |
Ruddy KJ, Partridge AH (2012) The unique reproductive concerns of young women with breast cancer. Adv Exp Med Biol 732: 77-87. doi: 10.1007/978-94-007-2492-1_6
|
| [18] |
Gomez-Campelo P, Bragado-Alvarez C, Hernandez-Lloreda MJ (2014) Psychological distress in women with breast and gynecological cancer treated with radical surgery. Psychooncology 23: 459-466. doi: 10.1002/pon.3439
|
| [19] |
Rosenberg SM, Tamimi RM, Gelber S, et al. (2014) Treatment-related amenorrhea and sexual functioning in young breast cancer survivors. Cancer 120: 2264-2271. doi: 10.1002/cncr.28738
|
| [20] |
Fontein DB, de Glas NA, Duijm M, et al. (2013) Age and the effect of physical activity on breast cancer survival: A systematic review. Cancer Treat Rev 39: 958-965. doi: 10.1016/j.ctrv.2013.03.008
|
| [21] |
Lan B, Jiang S, Li T, et al. (2020) Depression, anxiety, and their associated factors among Chinese early breast cancer in women under 35 years of age: A cross sectional study. Curr Probl Cancer 4: 100558. doi: 10.1016/j.currproblcancer.2020.100558
|
| [22] |
Mandelblatt JS, Zhai W, Ahn J, et al. (2020) Symptom burden among older breast cancer survivors: The Thinking and Living With Cancer (TLC) study. Cancer 126: 1183-1192. doi: 10.1002/cncr.32663
|
| [23] |
Champion VL, Wagner LI, Monahan PO, et al. (2014) Comparison of younger and older breast cancer survivors and age-matched controls on specific and overall quality of life domains. Cancer 120: 2237-2246. doi: 10.1002/cncr.28737
|
| [24] |
Sheppard VB, Harper FW, Davis K, et al. (2014) The importance of contextual factors and age in association with anxiety and depression in Black breast cancer patients. Psychooncology 23: 143-150. doi: 10.1002/pon.3382
|
| [25] |
Fried EI, Nesse RM, Zivin K, et al. (2014) Depression is more than the sum score of its parts: individual DSM symptoms have different risk factors. Psychol Med 44: 2067-2076. doi: 10.1017/S0033291713002900
|
| [26] |
Ten Have M, Lamers F, Wardenaar K, et al. (2016) The identification of symptom-based subtypes of depression: A nationally representative cohort study. J Affect Disord 190: 395-406. doi: 10.1016/j.jad.2015.10.040
|
| [27] |
Kroenke K, Spitzer RL, Williams JB (2001) The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 16: 606-613. doi: 10.1046/j.1525-1497.2001.016009606.x
|
| [28] |
Andersen BL, DeRubeis RJ, Berman BS, et al. (2014) Screening, assessment, and care of anxiety and depressive symptoms in adults with cancer: an American Society of Clinical Oncology guideline adaptation. J Clin Oncol 32: 1605-1619. doi: 10.1200/JCO.2013.52.4611
|
| [29] |
Nylund KL, Asparouhov T, Muthén BO (2007) Deciding on the Number of Classes in Latent Class Analysis and Growth Mixture Modeling: A Monte Carlo Simulation Study. Struct Equ Modeling 14: 535-569. doi: 10.1080/10705510701575396
|
| [30] |
Zhu L, Ranchor AV, van der Lee M, et al. (2016) Subtypes of depression in cancer patients: an empirically driven approach. Support Care Cancer 24: 1387-1396. doi: 10.1007/s00520-015-2919-y
|
| [31] |
Li J, Zhang H, Shao D, et al. (2019) Depressive Symptom Clusters and Their Relationships With Anxiety and Posttraumatic Stress Disorder Symptoms in Patients With Cancer: The Use of Latent Class Analysis. Cancer Nurs 42: 388-395. doi: 10.1097/NCC.0000000000000624
|
| [32] |
Hinz A, Mehnert A, Kocalevent RD, et al. (2016) Assessment of depression severity with the PHQ-9 in cancer patients and in the general population. BMC Psychiatry 16: 22. doi: 10.1186/s12888-016-0728-6
|
| [33] |
Kim JM, Jang JE, Stewart R, et al. (2013) Determinants of suicidal ideation in patients with breast cancer. Psychooncology 22: 2848-2856. doi: 10.1002/pon.3367
|
| [34] |
Dooley LN, Ganz PA, Cole SW, et al. (2016) Val66Met BDNF polymorphism as a vulnerability factor for inflammation-associated depressive symptoms in women with breast cancer. J Affect Disord 197: 43-50. doi: 10.1016/j.jad.2016.02.059
|
| [35] |
Carpenter JS, Igega CM, Otte JL, et al. (2014) Somatosensory amplification and menopausal symptoms in breast cancer survivors and midlife women. Maturitas 78: 51-55. doi: 10.1016/j.maturitas.2014.02.006
|
| [36] |
Howard-Anderson J, Ganz PA, Bower JE, et al. (2012) Quality of life, fertility concerns, and behavioral health outcomes in younger breast cancer survivors: a systematic review. J Natl Cancer Inst 104: 386-405. doi: 10.1093/jnci/djr541
|
| [37] |
Posner K, Brown GK, Stanley B, et al. (2011) The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168: 1266-1277. doi: 10.1176/appi.ajp.2011.10111704
|
Figures(3) / Tables(4)

Yanyan Li, Hong Liu, Yaoyao Sun, Jie Li, Yanhong Chen, Xuan Zhang, Juan Wang, Liuliu Wu, Di Shao, Fenglin Cao. Characteristics and subtypes of depressive symptoms in Chinese female breast cancer patients of different ages: a cross-sectional study[J]. AIMS Public Health, 2021, 8(4): 691-703. doi: 10.3934/publichealth.2021055







 DownLoad:
DownLoad: