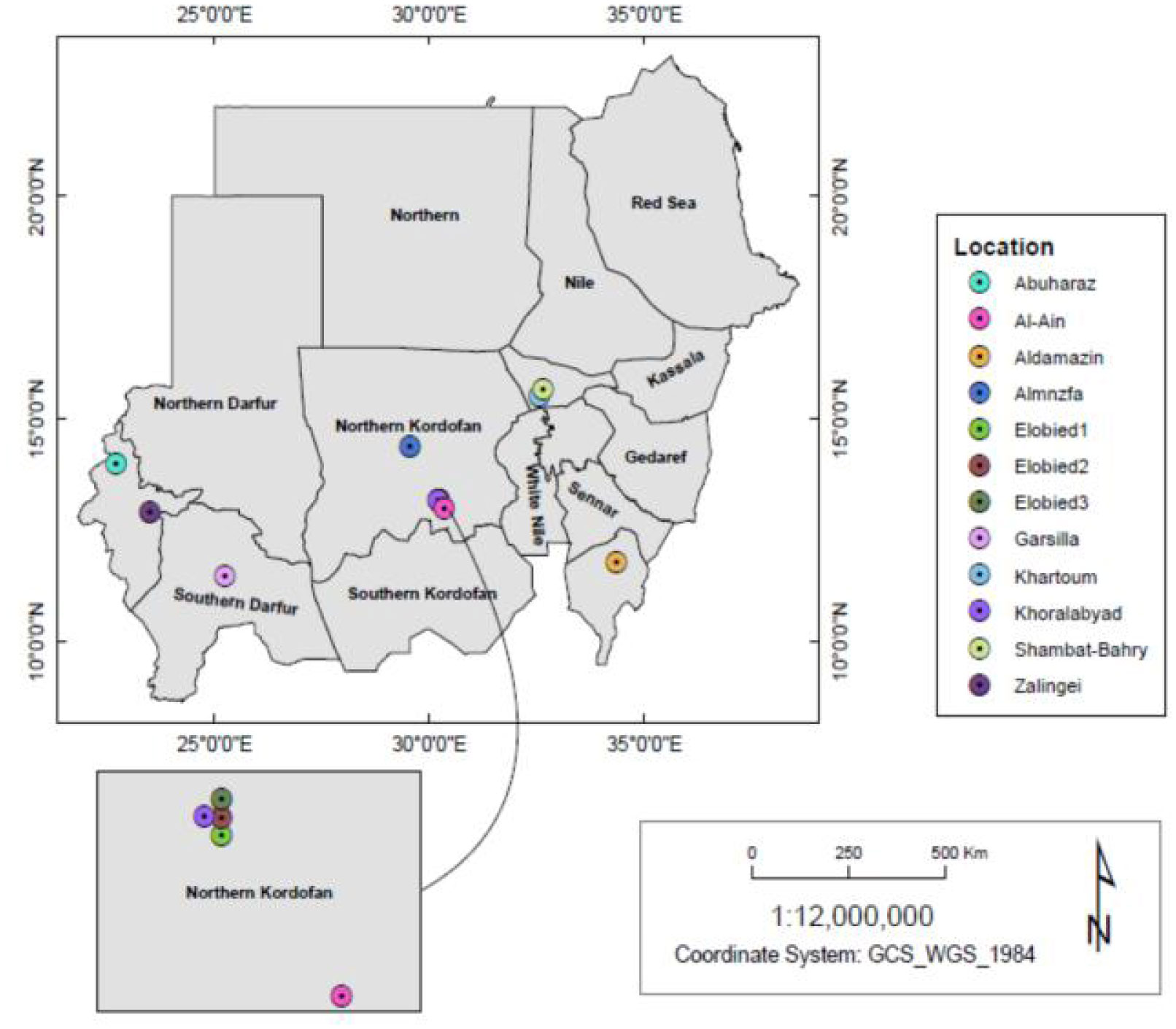
This research is conducted to study the genetic diversity of the genus Grewia tenax in Sudan. This plant has a common vernacular Sudanese Arabic name: “Gudeim”. It is mainly cultivated in North Darfur and Western Sudan. Common uses of Grewia species were overviewed in areas of nutrition, folk medicine and famine food. The genetic variation and genetic relationships among Grewia spp populations from different regions were efficiently determined using RAPD and ISSR markers.
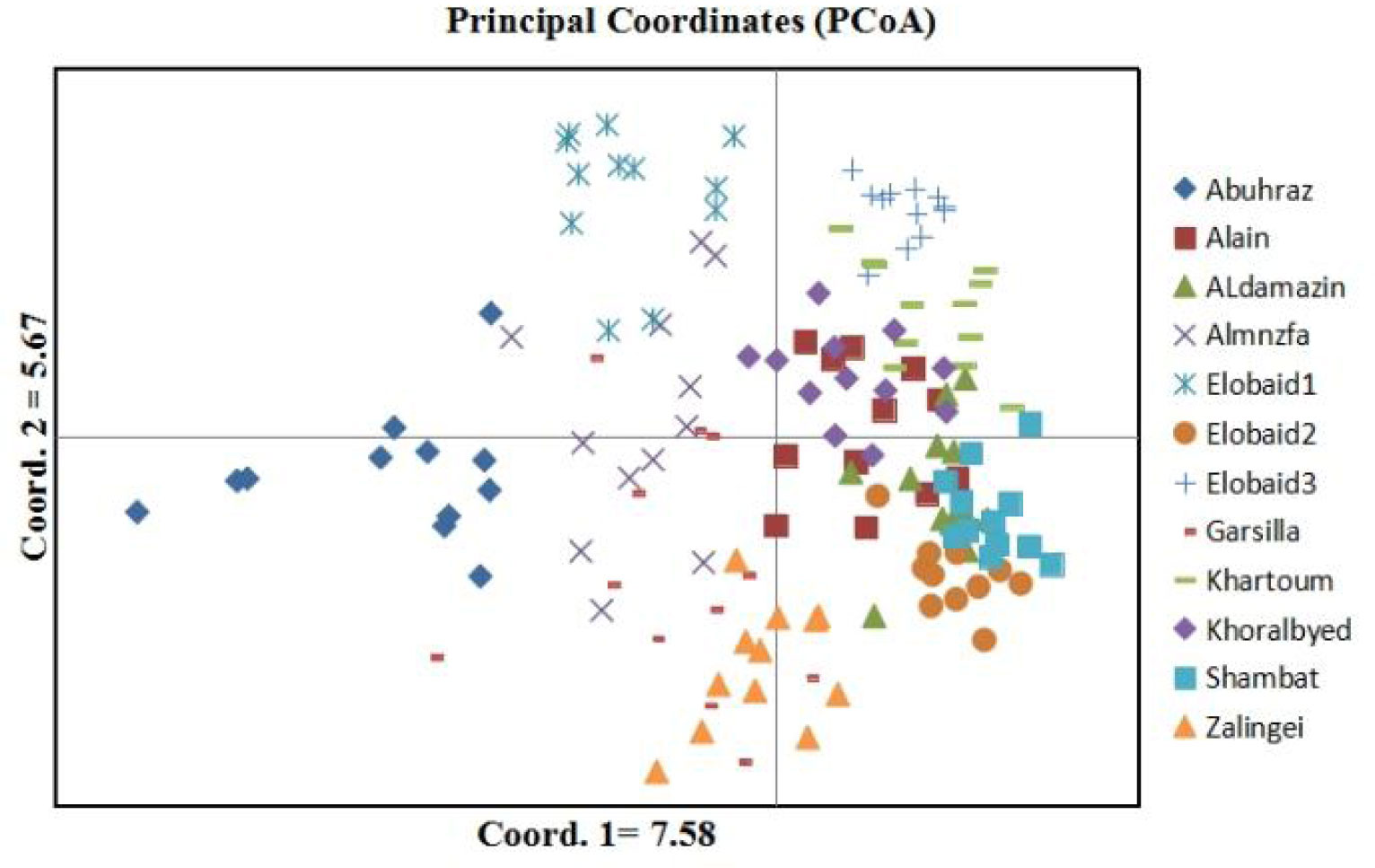
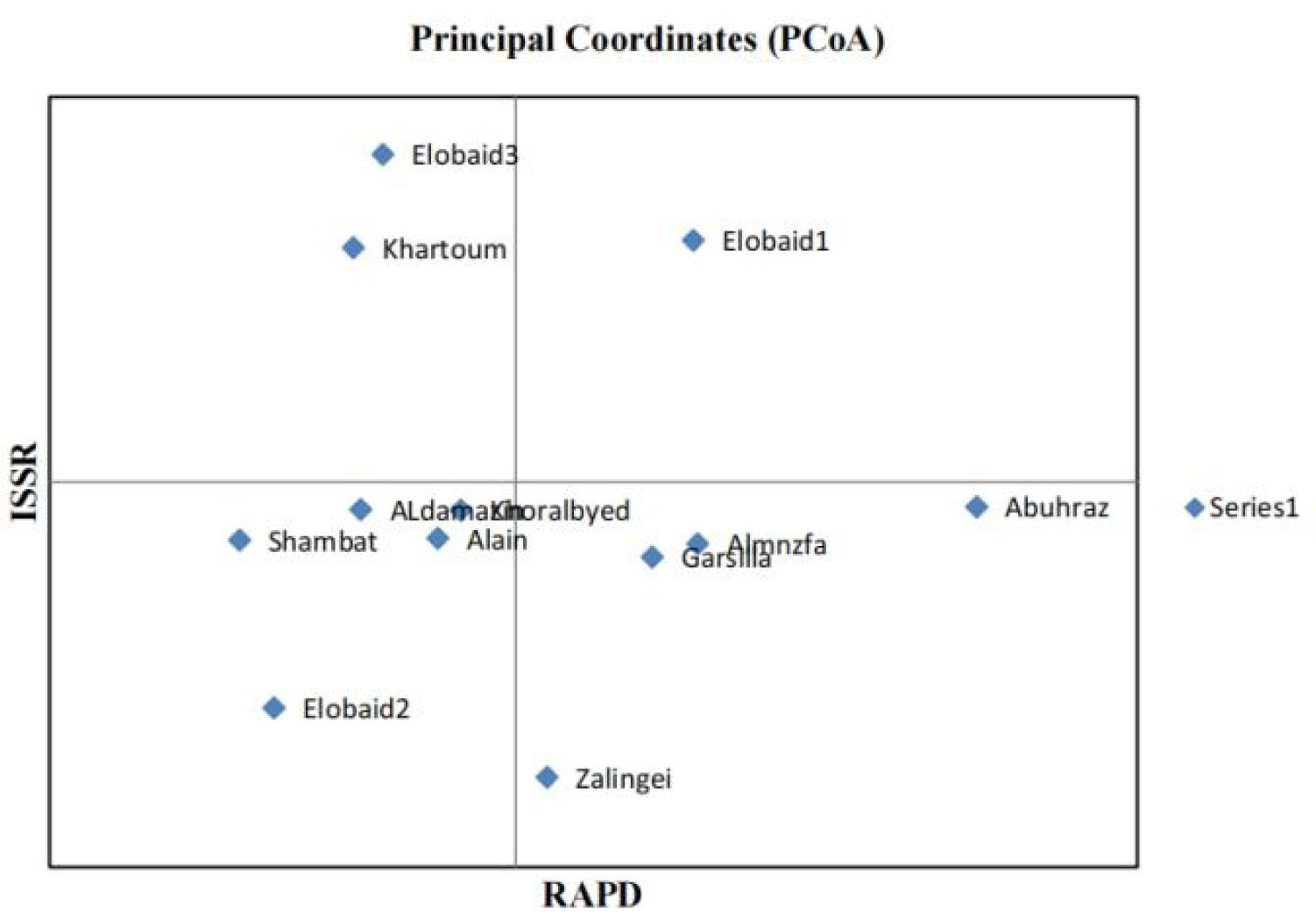
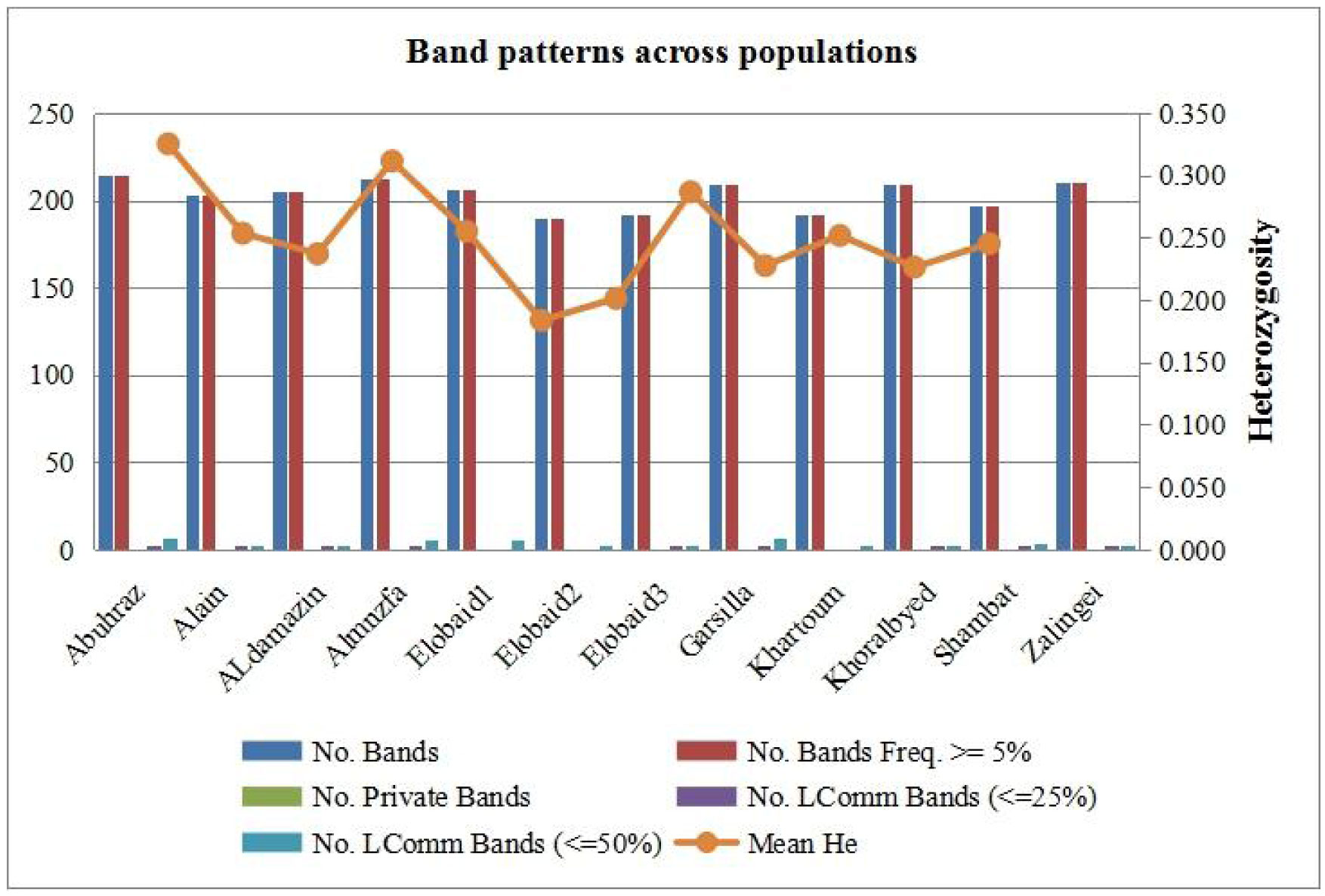
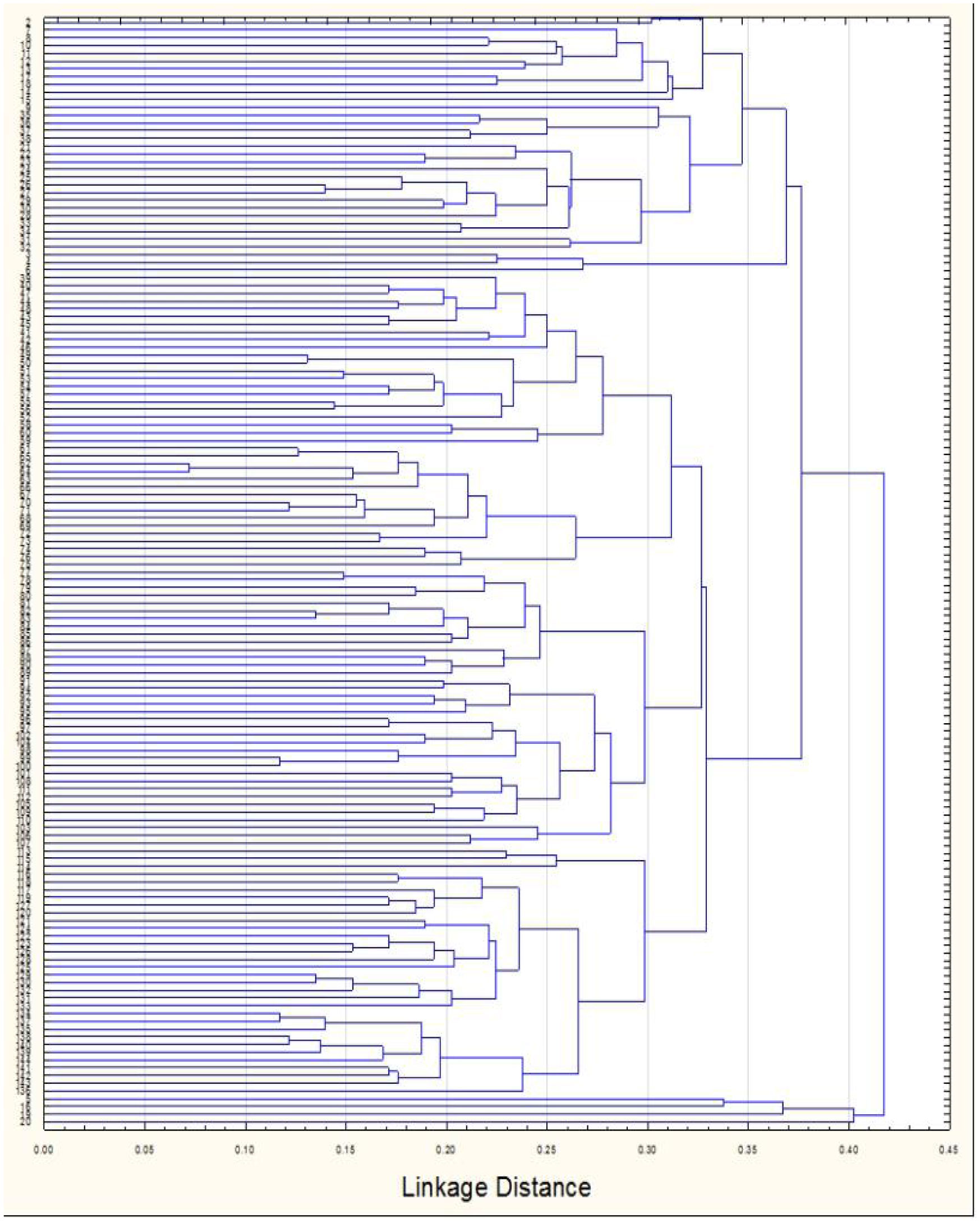
The results of genetic analysis were statistically analyzed by STATISTCA and GenALEx 6.5 software. The results of molecular variance revealed that 74% of total genetic diversity was due to within populations variations as opposed to 26% due to variations between populations unweighted pair- group method with arithmetic average (UPGMA) were constructed for and RAPD + ISSR. The UPGMA results showed variability of Grewia genotypes.
It was concluded that both the marker systems RAPD and ISSR combination can be effectively used in determination of genetic relationships among Grewia tenax genotypes.
Citation: Rami S. Habeballa, Mutasim Mohamed Khalafalla Ali, Rashied S. Modawi. Assessment of phylogenetic diversity and relationships among Grewia tenax population in Sudan using RAPD and ISSR molecular markers[J]. AIMS Molecular Science, 2022, 9(1): 1-24. doi: 10.3934/molsci.2022001
This research is conducted to study the genetic diversity of the genus Grewia tenax in Sudan. This plant has a common vernacular Sudanese Arabic name: “Gudeim”. It is mainly cultivated in North Darfur and Western Sudan. Common uses of Grewia species were overviewed in areas of nutrition, folk medicine and famine food. The genetic variation and genetic relationships among Grewia spp populations from different regions were efficiently determined using RAPD and ISSR markers.
The results of genetic analysis were statistically analyzed by STATISTCA and GenALEx 6.5 software. The results of molecular variance revealed that 74% of total genetic diversity was due to within populations variations as opposed to 26% due to variations between populations unweighted pair- group method with arithmetic average (UPGMA) were constructed for and RAPD + ISSR. The UPGMA results showed variability of Grewia genotypes.
It was concluded that both the marker systems RAPD and ISSR combination can be effectively used in determination of genetic relationships among Grewia tenax genotypes.

| [1] | Von M (1990) Trees and Shrubs of the Sahel: Their characteristics and uses. GTZ, Verlag Josef Margraf, Deutschland 31: 133-142. |
| [2] |
Gebauer J, El-Siddig K, El Tahir A, et al. (2007) Exploiting the potential of indigenous fruit trees: Grewia tenax (Forssk.) Fiori in Sudan. Genet Resour Crop Evol 54: 1701-1708. https://doi.org/10.1007/s10722-006-9178-1 
|
| [3] |
Rosa R, Melecchi M, Halmenschlager R, et al. (2006) Antioxidant and antimutagenic properties of Hibiscus tiliaceus L. methanolic extract. J Agric Food Chem 54: 7324-7330. https://doi.org/10.1021/jf061407b
|
| [4] | El Amin HM (1990) Trees and shrubs of the Sudan. Exeter, UK: Ithaca Press. |
| [5] | Sharma N, Patni V (2013) In vivo and in vitro qualitative phytochemical screening of Grewia species. Int j biol pharm res 9: 634-639. |
| [6] |
Gebauer J, El Siddig K, El Tahir BA, et al. (2007) Exploiting the potential of indigenous fruit trees. Grewia tenax (Forssk.) Fiori in Sudan. Genet Resour Crop Evol 54: 1701-1708. https://doi.org/10.1007/s10722-006-9178-1
|
| [7] | Mabry R (2009) Screening for iron deficiency anemia –including iron supplementation for children and pregnant women. Am Fam Physician 10: 897-898. |
| [8] |
Ballal ME, ElSiddig EA, Elfadl MA, et al. (2005) Gum arabic yield in differently managed Acacia senegal stands in western Sudan. Agrofor Syst 63: 237-245. https://doi.org/10.1007/s10457-005-4162-x
|
| [9] |
Khemis F, Ghoul M, Moshtaghie A, et al. (2006) Study of the effect of aqueous extract of Grewia tenax fruit on iron absorption by averted gut sac. J Ethnopharmacol 103: 90-98. https://doi.org/10.1016/j.jep.2005.07.017
|
| [10] |
Behbahani M, Shanehsazzadeh M, Hessami M (2011) Optimization of callus and cell suspension cultures of barring tonia racemosa (Lecythidaceae family) for lycopene production. Sci agric (Piracicaba, Braz) 68: 69-76. https://doi.org/10.1590/S0103-90162011000100011
|
| [11] | Ahmed ME, Hamid HB, Babikir HE, et al. (2012) Effects of Grewia tenax (Guddaim) as a natural food on the hemoglobin level and growth among displaced children of Darfur State, Western Sudan. J Med Med Sci 11: 729-733. |
| [12] | Weising K, Nybom H, Wolef K, et al. (2005) DNA fingerprinting in plants. Principles, Methods and Applications. Taylor and Francis Group 444. https://doi.org/10.1201/9781420040043 |
| [13] |
Hamrick L, Murawski A (1990) The breeding structure of tropical trees. Plant Species Biol 5: 157-165. https://doi.org/10.1111/j.1442-1984.1990.tb00200.x
|
| [14] |
Loveless D (1992) Isozyme variation in tropical trees: patterns of genetic organization. New Forest 6: 67-94. https://doi.org/10.1007/BF00120640
|
| [15] | Botstein D (1980) Construction of a genetic-linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 3: 314-31. |
| [16] |
Williams K, Kubelik R, Livak J, et al. (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18: 6531-6535. https://doi.org/10.1093/nar/18.22.6531
|
| [17] | Wolfe D, Liston A (1998) Contributions of PCR-based methods to plant systematics and evolutionary biology. Molecular systematics of plants: DNA sequencing 6: 4615-5419. https://doi.org/10.1007/978-1-4615-5419-6_2 |
| [18] |
Akkaya S, Bhagwat A, Cregan B (1992) Length polymorphisms of simple-sequence repeat DNA in soybean. Genetics 132: 1131-1139. https://doi.org/10.1093/genetics/132.4.1131
|
| [19] |
Vos P, Hogers R, Bleeker M, et al. (1995) AFLP a new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407-4414. https://doi.org/10.1093/nar/23.21.4407
|
| [20] | Sensi R, Vignani W, Rohde W, et al. (1996) Characterization of genetic biodiversity with Vitisviní feraL. Sangiovese and coloring genotypes by AFLP and ISTR, DNA marker technology. Vitis 35: 183-188. |
| [21] |
Saiki K (1988) Primer-directed enzymatic amplification of DNA with a thermo stable DNA polymerase. Science 239: 487-491. https://doi.org/10.1126/science.2448875
|
| [22] |
Dellaporta L, Wood J, Hicks B (1983) A plant DNA mini preparation: version II. Plant Mol Biol Rep 1: 19-21. https://doi.org/10.1007/BF02712670
|
| [23] | Sambrook J, Fritschi, Maniatis T (1989) Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press 82-88. |
| [24] | Archana V, Singh B, Saresh V, et al. (2015) RAPD and ISSR markers for molecular characterization of Grewia optiva: an important fodder tree of north western Himalayas. Range Management and Agroforestry 1: 2632. |
| [25] | Zuker M (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide, in RNA Biochemistry and biotechnology. Academic publishers 70: 11-43. https://doi.org/10.1007/978-94-011-4485-8_2 |
| [26] | Victoria M, Nour N, Jens R (2016) Gel staining Techniques-Dyeing to know it all. Wiley online library 1-10. |
| [27] |
Nei M, Li W (1979) Mathematical model for studying genetic variation in terms of restriction endonuclease. Proc Natl Acad Sci 76: 5269-5273. https://doi.org/10.1073/pnas.76.10.5269
|
| [28] | Rohlf F (1993) NTSYS-pc. Numerical taxonomy and multivariate analysis system. Version 1.18. New York: Exeter: Setauket. |
| [29] |
Peakall R, Smouse E (2006) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28: 2537-2539. https://doi.org/10.1093/bioinformatics/bts460
|
| [30] |
Chase R, Boshier H, Bawa S (1995) population genetics of Cordia alliodora (Boraginaceae), a Neoytopical tree. Genetic variation in natural population. Am J Bot 4: 468-475. https://doi.org/10.1002/j.1537-2197.1995.tb15666.x
|
| [31] | R M (1995) Neutrality tests for molecular markers and the connection between DNA polymorphism, demography, and conservation biology. Biol Conserv 10: 665-671. https://doi.org/10.1046/j.1523-1739.1996.10020665.x |
| [32] |
Ford J (2002) Application of selective neutrality tests to molecular ecology. Mol Ecol Notes 11: 1245-1262. https://doi.org/10.1046/j.1365-294X.2002.01536.x
|
| [33] |
Van O, Hutchinson F, Wills M, et al. (2003) Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 3: 535-538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
|
| [34] |
Divaret I, Margale E, Thomas, G (1999) RAPD markers on seed bulks efficiently assess the genetic diversity of a Brassica oleracea L. collection. Theor Appl Genet 98: 1029-1035. https://doi.org/10.1007/s001220051164
|
| [35] |
Kermer A, Caron H, Cavers S, et al. (2005) Monitoring genetic diversity in tropical trees with multi locus dominant markers. Heredity 95: 274-280. https://doi.org/10.1038/sj.hdy.6800738
|
| [36] |
Martins P, Lima J, Gomes S, et al. (2007) RAPD and ISSR molecular markersin Olea europaea L. genetic variability and molecular cultivar identification. Genet Resour Crop Evol 54: 117-128. https://doi.org/10.1007/s10722-005-2640-7
|
| [37] |
Ngezahayo F, Dong Y, Liu B (2007) Soma clonal variation at the nucleotide sequence level in rice (Oryza sativa L.) as revealed by RAPD and ISSR markers, and by pairwise sequence analysis. J Appl Genet 48: 329-336. https://doi.org/10.1007/BF03195229
|
| [38] |
Maslova V (2008) Differentiation of two hemp nettle species (Galeopsis bifida Boenn. and G. tetrahit L.) inferred from morphological characters and DNA markers. Russ J Genet 44: 312-319. https://doi.org/10.1134/S1022795408030113
|
| [39] | Mohanty S, Das B, Ghosh N, et al. (2010) Genetic diversity of 28 wild species of fodder legume Cassia using RAPD, ISSR, and SSR markers: a novel breeding strategy. J Biotech Res 2: 44-55. |
| [40] |
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Biol Conserv 10: 1500-1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
|
| [41] | Karp A, Kresovich S, Bha K, et al. (1997) Molecular tools in plant genetic resources conservation: A Guide to the technologies, IPGRI Technical Bulletin No. 2. International Plant Genetic Resources Institute : 3-17. |
| [42] |
Zietkiewicz E, Rafalshi A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomic 20: 176-183. https://doi.org/10.1006/geno.1994.1151
|
| [43] | Wolf D, Randle P (2001) Relationships within and among species of the holoparasitic genus Hyobanche (Orobanchaceae) inferred from ISSR banding patterns and nucleotide sequences. Syst Bot 26: 120-130. |
| [44] |
Li A, Ge S (2001) Genetic variation and clonal diversity of Psammochloa villosa (Poaceae) detected by ISSR markers. Ann Bot 87: 585-590. https://doi.org/10.1006/anbo.2001.1390
|
| [45] |
Pazouki L, Mardi M, Shanjani S, et al. (2010) Genetic diversity and relationships among Pistacia species and cultivars. Conserv Genet 11: 311-318. https://doi.org/10.1007/s10592-009-9812-5
|
| [46] | Baghizadeh A, Noroozi S, Javaran J (2010) Study on genetic diversity of some Iranian pistachio(Pistacia vera L.) cultivars using random amplified polymorphic DNA (RAPD), inter simple sequence repeat (ISSR) and simple sequence repeat (SSR) markers: a comparative study. Afr j biotechnol 45: 7632-7640. |
| [47] |
Ahmad R, Ferguson L, Southwick SM (2003a) Identification of pistachio (Pistacia vera L.) nuts with microsatellite markers. J Am Soc Hortic Sci 128: 898-903. https://doi.org/10.21273/JASHS.128.6.0898
|
| [48] |
Chhajer S, Kalia R (2016) Evaluation of genetic homogeneity in vitro-raised plants of Tecomella undulata (Sm.) Seem. using molecular markers. Tree Genet Genomes 12: 83-100. https://doi.org/10.1007/s11295-016-1057-0
|
| [49] |
Morgante M, Olivieri M (1993) PCR-amplified microsatellites as markers in plant genetics. Plant J 3: 175-182. https://doi.org/10.1111/j.1365-313X.1993.tb00020.x
|
| [50] |
Depeiges A, Goubely C, Lenoir A, et al. (1995) Identification of the most represented motifs in Arabidopsis thaliana microsatellite loci. Theor Appl Genet 91: 160-168. https://doi.org/10.1007/BF00220873
|
| [51] | Blair W, Panaud O, McCouch R (1999) Inter-simple sequences repeat (ISSR) amplification for analysis of microsatellite motif frequency and fingerprinting in rice (Oryza sativa L.). Inte Rice Res 98: 780-792. https://doi.org/10.1007/s001220051135 |
| [52] |
Casasoli M, Mattioni C, Cherubini M, et al. (2001) Genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR, and isozyme markers. Theor Appl Genet 102: 1190-1199. https://doi.org/10.1007/s00122-001-0553-1
|
| [53] |
Moreno S, Martin P, Oritz M (1998) Inter-simple sequence repeat PCR for characterization of closely related grapevine germplasm. Euphytica 101: 117-125. https://doi.org/10.1023/A:1018379805873
|
| [54] |
Nagaoka T, Ogihara Y (1997) Applicability of inter-simple sequence repeat polymorphisms in wheat for use as DNA markers in comparison to RFLP and RAPD markers. Theor Appl Genet 94: 597-602. https://doi.org/10.1007/s001220050456
|
| [55] |
Verma K, Lal S, Ahmed N, et al. (2015) Genetic diversity among native wild hip rose (Rosa canina L.) genotypes collected from Kashmir valley. Indian J Hortic 72: 250-256. https://doi.org/10.5958/0974-0112.2015.00047.X
|
| [56] | Srivastava P, Kunjupillai A, Awasthi K, Saratchandra B (2004) genetic analysis of Morusalba. Indian J Biotechnol 3: 527-532. |
| [57] |
Sharma K, Kumar S, Rawat D, et al. (2011) Genetic diversity and gene flow estimation in Prosopis cineraria (L.) Druce: a key stone tree species of Indian Thar desert. Biochem Syst Ecol 39: 9-13. https://doi.org/10.1016/j.bse.2010.12.018
|
| [58] | Josiah C, George O, Eleazar M, et al. (2008) Genetic diversity in Kenyan populations of Acacia senegal (L.) willd revealed by combined RAPD and ISSR markers. Afr J Biotechnol 7: 2333-2340. |
| [59] | Yorgancilar M., Babaoglu E, Hakki E., et al. (2009) Determination of the relationship among oldworld lupin (Lupinu ssp.) species using RAPD and ISSR markers. Afr J Biotechnol 8: 3524-3530. |
| [60] |
Gupta PK, Rustgi S, Mir RR (2008) Array-Based High-Throughput DNA Markers for Crop Improvement. Heredity 101: 5-18. https://doi.org/10.1038/hdy.2008.35
|
| [61] |
Tripathi M, Tyagi A, Kumar A, et al. (2013) The internal transcribed spacer (ITS) region and trnH-psbA [corrected] are suitable candidate loci for DNA barcoding of tropical tree species of India. PLoS One 8: 57934. https://doi.org/10.1371/journal.pone.0057934
|
| [62] | Mukherjee A, Sikdar B, Ghosh B, et al. (2013) RAPD and ISSR analysis of some economically important species, varieties, and cultivars of the genus Allium (Alliaceae). Turk J Bot 37: 605-618. https://doi.org/10.3906/bot-1208-18 |
Figures(5) / Tables(7)

Rami S. Habeballa, Mutasim Mohamed Khalafalla Ali, Rashied S. Modawi. Assessment of phylogenetic diversity and relationships among Grewia tenax population in Sudan using RAPD and ISSR molecular markers[J]. AIMS Molecular Science, 2022, 9(1): 1-24. doi: 10.3934/molsci.2022001







 DownLoad:
DownLoad: