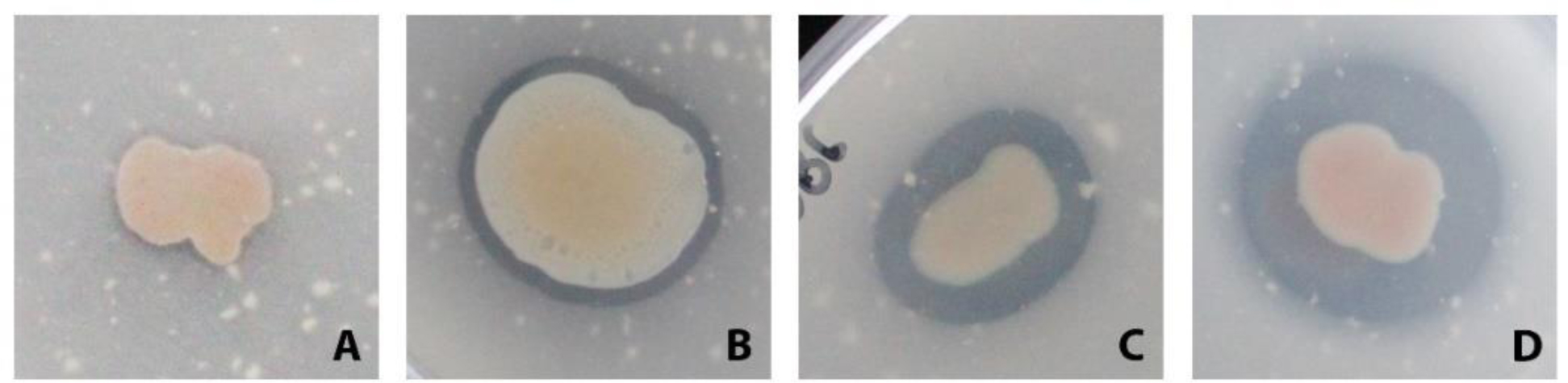
Plastics have quickly become an integral part of modern life. Due to excessive production and improper waste disposal, they are recognized as contaminants present in practically all habitat types. Although there are several polymers, polyethylene terephthalate (PET) is of particular concern due to its abundance in the environment. There is a need for a solution that is both cost-effective and ecologically friendly to address this pollutant. The use of microbial depolymerizing enzymes could offer a biological avenue for plastic degradation, though the full potential of these enzymes is yet to be uncovered. The purpose of this study was to use (1) plate-based screening methods to investigate the plastic degradation potential of marine bacteria from the order Enterobacterales collected from various organismal and environmental sources, and (2) perform genome-based analysis to identify polyesterases potentially related to PET degradation. 126 bacterial isolates were obtained from the strain collection of RD3, Research Unit Marine Symbioses-GEOMAR-and sequentially tested for esterase and polyesterase activity, in combination here referred to as PETase–like activity. The results show that members of the microbial families Alteromonadaceae, Shewanellaceae, and Vibrionaceae, derived from marine sponges and bryozoans, are the most promising candidates within the order Enterobacterales. Furthermore, 389 putative hydrolases from the α/β superfamily were identified in 23 analyzed genomes, of which 22 were sequenced for this study. Several candidates showed similarities with known PETases, indicating underlying enzymatic potential within the order Enterobacterales for PET degradation.
Citation: Denisse Galarza–Verkovitch, Onur Turak, Jutta Wiese, Tanja Rahn, Ute Hentschel, Erik Borchert. Bioprospecting for polyesterase activity relevant for PET degradation in marine Enterobacterales isolates[J]. AIMS Microbiology, 2023, 9(3): 518-539. doi: 10.3934/microbiol.2023027
Plastics have quickly become an integral part of modern life. Due to excessive production and improper waste disposal, they are recognized as contaminants present in practically all habitat types. Although there are several polymers, polyethylene terephthalate (PET) is of particular concern due to its abundance in the environment. There is a need for a solution that is both cost-effective and ecologically friendly to address this pollutant. The use of microbial depolymerizing enzymes could offer a biological avenue for plastic degradation, though the full potential of these enzymes is yet to be uncovered. The purpose of this study was to use (1) plate-based screening methods to investigate the plastic degradation potential of marine bacteria from the order Enterobacterales collected from various organismal and environmental sources, and (2) perform genome-based analysis to identify polyesterases potentially related to PET degradation. 126 bacterial isolates were obtained from the strain collection of RD3, Research Unit Marine Symbioses-GEOMAR-and sequentially tested for esterase and polyesterase activity, in combination here referred to as PETase–like activity. The results show that members of the microbial families Alteromonadaceae, Shewanellaceae, and Vibrionaceae, derived from marine sponges and bryozoans, are the most promising candidates within the order Enterobacterales. Furthermore, 389 putative hydrolases from the α/β superfamily were identified in 23 analyzed genomes, of which 22 were sequenced for this study. Several candidates showed similarities with known PETases, indicating underlying enzymatic potential within the order Enterobacterales for PET degradation.

| [1] |
Carr C, de Oliveira B, Jackson S, et al. (2022) Identification of BgP, a cutinase-like polyesterase from a deep-sea sponge-derived Actinobacterium. Front Microbiol 13: 888343. https://doi.org/10.3389/fmicb.2022.888343 
|
| [2] |
Dalmaso G, Ferreira D, Vermelho A (2015) Marine extremophiles: a source of hydrolases for biotechnological applications. Mar Drugs 13: 1925-1965. https://doi.org/10.3390/md13041925
|
| [3] |
Rosenberg E, Zilber-Rosenberg I (2018) The hologenome concept of evolution after 10 years. Microbiome 6: 1-14. https://doi.org/10.1186/s40168-018-0457-9
|
| [4] |
de Oliveira B, Carr C, Dobson A, et al. (2020) Harnessing the sponge microbiome for industrial biocatalysts. Appl Microbiol Biotechnol 10: 8131-8154. https://doi.org/10.1007/s00253-020-10817-3
|
| [5] |
Arnosti C, Bell C, Moorhead D, et al. (2014) Extracellular enzymes in terrestrial, freshwater, and marine environments: perspectives on system variability and common research needs. Biogeochemistry 117: 5-21. https://doi.org/10.1007/s10533-013-9906-5
|
| [6] | PlasticsEuropePlastics–The Facts 2022: An Analysis of European plastics production, demand, and waste (2023). Available from: https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/. |
| [7] |
Jambeck J, Geyer R, Wilcox C, et al. (2015) Plastic waste inputs from land into the ocean. Science 347: 768-771. https://doi.org/10.1126/science.1260352
|
| [8] |
Oberbeckmann S, Osborn A, Duhaime M (2016) Microbes on a bottle: substrate, season and geography influence community composition of microbes colonizing marine plastic debris. PLoS One 11: e0159289. https://doi.org/10.1371/journal.pone.0159289
|
| [9] |
Cox K, Covernton G, Davies H, et al. (2019) Human consumption of microplastics. Environ Sci Technol 53: 7068-7074. https://doi.org/10.1021/acs.est.9b01517
|
| [10] |
Gregory M (2009) Environmental implications of plastic debris in marine settings entanglement, ingestion, smothering, hangers-on, hitch-hiking and alien invasions. Phil Trans R Soc B 364: 2013-2025. https://doi.org/10.1098/rstb.2008.0265
|
| [11] |
Worm B, Lotze H, Jubinville I, et al. (2017) Plastic as a persistent marine pollutant. Annu Rev Environ Resour 42: 1-26. https://doi.org/10.1146/annurev-environ-102016-060700
|
| [12] |
Carlton J, Fowler A (2018) Ocean rafting and marine debris: A broader vector menu requires a greater appetite for invasion biology research support. Aquat Invasions 13: 11-15. https://doi.org/10.3391/ai.2018.13.1.02
|
| [13] |
Tetu S, Sarker I, Moore L (2020) How will marine plastic pollution affect bacterial primary producers?. Commun Biol 3: 1-4. https://doi.org/10.1038/s42003-020-0789-4
|
| [14] |
Bowley J, Baker–Austin C, Porter A, et al. (2021) Oceanic hitchhikers–assessing pathogen risks from marine microplastic. Trends Microbiol 29: 107-116. https://doi.org/10.1016/j.tim.2020.06.011
|
| [15] |
Pirillo V, Pollegioni L, Molla G (2021) Analytical methods for the investigation of enzyme–catalyzed degradation of polyethylene terephthalate. FEBS J 288: 4730-4745. https://doi.org/10.1111/febs.15850
|
| [16] |
Koshti R, Mehta L, Samarth N (2018) Biological recycling of polyethylene terephthalate: A mini–review. J Polym Environ 26: 3520-3529. https://doi.org/10.1007/s10924-018-1214-7
|
| [17] | Zimmermann W (2019) Biocatalytic recycling of polyethylene terephthalate plastic. Philos Trans R Soc A: 378. https://doi.org/10.1098/rsta.2019.0273 |
| [18] |
Dissanayake L, Jayakody L (2021) Engineering microbes to bio-upcycle Polyethylene Terephthalate. Front Bioeng Biotechnol 9: 656465. https://doi.org/10.3389/fbioe.2021.656465
|
| [19] |
Tiso T, Narancic T, Wei R, et al. (2021) Towards bio-upcycling of polyethylene terephthalate. Metab Eng 66: 167-178. https://doi.org/10.1016/j.ymben.2021.03.011
|
| [20] |
Danso D, Chow J, Streit W (2019) Plastics: Environmental and biotechnological perspectives on microbial degradation. Appl Environ Microbiol 85: 01019-010195. https://doi.org/10.1128/AEM.01095-19
|
| [21] |
Schmidt J, Wei R, Oeser T (2017) Degradation of polyester polyurethane by bacterial polyester hydrolases. Polymers 9: 65. https://doi.org/10.3390/polym9020065
|
| [22] |
Carr C, Clarke D, Dobson A (2020) Microbial polyethylene terephthalate hydrolases: current and future perspectives. Front Microbiol 11: 1-23. https://doi.org/10.3389/fmicb.2020.571265
|
| [23] |
Mohanan N, Montazer Z, Sharma P, et al. (2020) Microbial and enzymatic degradation of synthetic plastics. Front Microbiol 11: 580709. https://doi.org/10.3389/fmicb.2020.580709
|
| [24] |
Ru J, Huo Y, Yang Y (2020) Microbial degradation and valorization of plastic wastes. Front Microbiol 11: 442. https://doi.org/10.3389/fmicb.2020.00442
|
| [25] |
Kawai F, Kawabata T, Oda M (2019) Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl Microbiol Biotechnol 103: 4253-4268. https://doi.org/10.1007/s00253-019-09717-y
|
| [26] |
Danso D, Schmeisser C, Chow J, et al. (2018) New insights into the function and global distribution of polyethylene terephthalate (PET)-degrading bacteria and enzymes in marine and terrestrial metagenomes. Appl Environ Microbiol 84: e02773-17. https://doi.org/10.1128/AEM.02773-17
|
| [27] |
Müller R, Schrader H, Profe J, et al. (2005) Enzymatic degradation of poly (ethylene terephthalate): Rapid Hydrolysis using a hydrolase from T. fusca. Macromol Rapid Commun 26: 1400-1405. https://doi.org/10.1002/marc.200500410
|
| [28] |
Yoshida S, Hiraga K, Takehana T, et al. (2016) A bacterium that degrades and assimilates poly (ethylene terephthalate). Science 351: 1196-1199. https://doi.org/10.1126/science.aad6359
|
| [29] |
Ekanayaka A, Tibpromma S, Dai D, et al. (2022) A review of the fungi that degrade plastic. J Fungi 8: 772. https://doi.org/10.3390/jof8080772
|
| [30] |
Roth C, Wei R, Oeser T, et al. (2014) Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl Microbiol Biotechnol 98: 7815-7823. https://doi.org/10.1007/s00253-014-5672-0
|
| [31] |
Sulaiman S, Yamato S, Kanaya E, et al. (2012) Isolation of a novel cutinase homolog with polyethylene terephthalate–degrading activity from leaf–branch compost by using a metagenomic approach. Appl Environ Microbiol 78: 1556-1562. https://doi.org/10.1128/AEM.06725-11
|
| [32] |
Then J, Wei R, Oeser T, et al. (2016) A disulfide bridge in the calcium binding site of a polyester hydrolase increases its thermal stability and activity against polyethylene terephthalate. FEBS Open Bio 6: 425-432. https://doi.org/10.1002/2211-5463.12053
|
| [33] |
Gambarini V, Pantos O, Kingsbury J, et al. (2021) Phylogenetic distribution of plastic-degrading microorganisms. mSystems 6: e01112-20. https://doi.org/10.1128/mSystems.01112-20
|
| [34] |
Imhoff J (2005) Enterobacteriales. Bergey's manual® of systematic bacteriology . Springer 587-850. https://doi.org/10.1007/0-387-28022-7_13
|
| [35] | Brenner D, Farmer J (2005) III. Family Enterobacteriaceae. Bergey's manual® of systematic bacteriology . Springer 587-606. https://doi.org/10.1002/9781118960608.fbm00222 |
| [36] |
Janda J, Abbott S (2021) The changing face of the family Enterobacteriaceae (Order: “Enterobacterales”): New members, taxonomic issues, geographic expansion, and new diseases and disease syndromes. Clin Microbiol Rev 34: 1-45. https://doi.org/10.1128/CMR.00174-20
|
| [37] |
Kato C, Honma A, Sato S, et al. (2019) Poly 3-hydroxybutyrate-co- 3-hydroxyhexanoate films can be degraded by the deep-sea microbes at high pressure and low temperature conditions. High Press Res 39: 248-257. https://doi.org/10.1080/08957959.2019.1584196
|
| [38] |
Borchert E, García-Moyano A, Sanchez-Carrillo S, et al. (2021) Deciphering a marine bone-degrading microbiome reveals a complex community effort. mSystems 6: 1218-20. https://doi.org/10.1128/mSystems.01218-20
|
| [39] |
Yang J, Yang Y, Wu W, et al. (2014) Evidence of polyethylene biodegradation by bacterial strains from the guts of plastic-eating waxworms. Environ Sci Technol 48: 13776-13784. https://doi.org/10.1021/es504038a
|
| [40] |
Mallakuntla M, Vaikuntapu P, Bhuvanachandra B, et al. (2017) Transglycosylation by a chitinase from Enterobacter cloacae subsp. cloacae generates longer chitin oligosaccharides. Sci Rep 7: 1-12. https://doi.org/10.1038/s41598-017-05140-3
|
| [41] |
Volova T, Boyandin A, Vasil'ev A, et al. (2011) Biodegradation of polyhydroxyalkanoates (PHAs) in the South China Sea and identification of PHA-degrading bacteria. Microbiology 80: 252-260. https://doi.org/10.1134/S0026261711020184
|
| [42] |
Dashti N, Ali N, Eliyas M, et al. (2015) Most hydrocarbonoclastic bacteria in the total environment are diazotrophic, which highlights their value in the bioremediation of hydrocarbon contaminants. Microbes Environ 30: 70-75. https://doi.org/10.1264/jsme2.ME14090
|
| [43] |
Wang X, Isbrandt T, Strube M, et al. (2021) Chitin degradation machinery and secondary metabolite profiles in the marine bacterium Pseudoalteromonas rubra S4059. Mar Drugs 19: 108. https://doi.org/10.3390/md19020108
|
| [44] |
Caruso G (2020) Microbial colonization in marine environments: overview of current knowledge and emerging research topics. J Mar Sci Eng 8: 78. https://doi.org/10.3390/jmse8020078
|
| [45] |
Sekiguchi T, Saika A, Nomura K, et al. (2011) Biodegradation of aliphatic polyesters soaked in deep seawaters and isolation of poly(ϵ-caprolactone)-degrading bacteria. Poly. Degrad Stabil 96: 1397-1403. https://doi.org/10.1016/j.polymdegradstab.2011.03.004
|
| [46] |
Ohta Y, Hatada Y, Miyazaki M, et al. (2005) Purification and characterization of a novel α-agarase from a Thalassomonas sp. Curr Microbiol 50: 212-216. https://doi.org/10.1007/s00284-004-4435-z
|
| [47] |
Yakimov M, Bargiela R, Golyshin P (2022) Calm and Frenzy: marine obligate hydrocarbonoclastic bacteria sustain ocean wellness. Curr Opin Biotechnol 73: 337-345. https://doi.org/10.1016/j.copbio.2021.09.015
|
| [48] |
Molitor R, Bollinger A, Kubicki S, et al. (2020) Agar platebased screening methods for the identification of polyester hydrolysis by Pseudomonas species. Microb Biotechnol 13: 274-284. https://doi.org/10.1111/1751-7915.13418
|
| [49] |
Pérez-García P, Danso D, Zhang H, et al. (2021) Exploring the global metagenome for plastic-degrading enzymes. Methods Enzymol 648: 137-157. https://doi.org/10.1016/bs.mie.2020.12.022
|
| [50] | Genome Taxonomy Database (GTDB)University of Queensland (2023). Available from: https://gtdb.ecogenomic.org/. |
| [51] | R Studio Team (2020). Available from: https://support--rstudio-com.netlify.app/. |
| [52] |
Noda T, Sagara H, Yen A, et al. (2006) Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 439: 490-492. https://doi.org/10.1038/nature04378
|
| [53] |
Reysenbach A, Longnecker K, Kirshtein J (2000) Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a mid-atlantic ridge hydrothermal vent. Appl Environ Microbiol 66: 3798-3806. https://doi.org/10.1128/AEM.66.9.3798-3806.2000
|
| [54] |
Kumar S, Stecher G, Li M, et al. (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35: 1547-1549. https://doi.org/10.1093/molbev/msy096
|
| [55] |
Larkin M, Blackshields G, Brown N, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947-2948. https://doi.org/10.1093/bioinformatics/btm404
|
| [56] |
Price M, Dehal P, Arkin A (2009) Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641-1650. https://doi.org/10.1093/molbev/msp077
|
| [57] |
Price M, Dehal P, Arkin A (2010) FastTree 2-Approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490. https://doi.org/10.1371/journal.pone.0009490
|
| [58] | ITOL, Interactive Tree Of Life. Available from: https://itol.embl.de. |
| [59] |
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49: 293-296. https://doi.org/1093/nar/gkab301
|
| [60] | Desjardins P, Conklin D (2010) NanoDrop microvolume quantitation of nucleic acids. J Vis Exp 45: e2565. https://doi.org/10.3791/2565 |
| [61] |
Kolmogorov M, Yuan J, Lin Y, et al. (2019) Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540-546. https://doi.org/10.1038/s41587-019-0072-8
|
| [62] |
Parks D, Chuvochina M, Waite D, et al. (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36: 996-1004. https://doi.org/10.1038/nbt.4229
|
| [63] |
Chaumeil P, Mussig A, Hugenholtz P, et al. (2019) GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36: 1925-1927. https://doi.org/10.1093/bioinformatics/btz848
|
| [64] |
Hyatt D (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119-119. https://doi.org/10.1186/1471-2105-11-119
|
| [65] |
Eddy S (2011) Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195. https://doi.org/10.1371/journal.pcbi.1002195
|
| [66] |
Matsen F, Kodner R, Armbrust E (2010) pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 538. https://doi.org/10.1186/1471-2105-11-538
|
| [67] |
Huerta-Cepas J, Szklarczyk D, Heller D, et al. (2019) Eggnog 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47: 309-314. https://doi.org/nar/gky1085
|
| [68] |
Almeida E, Carrillo Rincón A, Jackson S, et al. (2019) In silico screening and heterologous expression of a polyethylene terephthalate hydrolase (PETase)-like enzyme (SM14est) with polycaprolactone (PCL)-degrading activity, from the marine sponge-derived strain Streptomyces sp. SM14. Front Microbiol 10: 2187. https://doi.org/10.3389/fmicb.2019.02187
|
| [69] |
Buchholz P, Feuerriegel G, Zhang H, et al. (2022) Plastics degradation by hydrolytic enzymes: The plastics-active enzymes database—PAZy. Proteins: Structure, Function, and Bioinformatics. Proteins 90: 1443-1456. https://doi.org/10.1002/prot.26325
|
| [70] |
Di Tommaso P, Moretti S, Xenarios I, et al. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39: 13-17. https://doi.org/10.1093/nar/gkr245
|
| [71] |
Gouet P, Courcelle E, Stuart D, et al. (1999) Espript: analysis of multiple sequence alignments in Postscript. Bioinformatics 15: 305-308. https://doi.org/10.1093/bioinformatics/15.4.305
|
| [72] | The Lipase Engineering Database (LED) version4.1.0. Germany: University of Stuttgart. Available from: https://led.biocatnet.de/. |
| [73] |
Armenteros J, Tsirigos K, Sønderby C, et al. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37: 420-423. https://doi.org/10.1038/s41587-019-0036-z
|
| [74] |
Baek M, DiMaio F, Anishchenko I, et al. (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373: 871-876. https://doi.org/10.1126/science.abj8754
|
| [75] |
Blázquez-Sánchez P, Engelberger F, Cifuentes-Anticevic J, et al. (2022) Antarctic polyester hydrolases degrade aliphatic and aromatic polyesters at moderate temperatures. Appl Environ Microbiol 88: e0184221. https://doi.org/10.1128/AEM.01842-21
|
| [76] |
Bollinger A, Thies S, Knieps-Grünhagen E, et al. (2020) A novel polyester hydrolase from the marine bacterium Pseudomonas aestusnigri–structural and functional insights. Front Microbiol 11: 1-16. https://doi.org/10.3389/fmicb.2020.00114
|
| [77] |
Ronkvist Å, Xie W, Lu W, et al. (2009) Cutinase-catalyzed hydrolysis of poly (ethylene terephthalate). Macromolecules 42: 5128-5138. https://doi.org/10.1021/ma9005318
|
| [78] | Eiamthong B, Meesawat P, Wongsatit T, et al. (2022) Discovery and genetic code expansion of a polyethylene terephthalate (PET) hydrolase from the human saliva metagenome for the degradation and bio-functionalization of PET. Angew Chem 61: e202203061. https://doi.org/10.1002/anie.202203061 |
| [79] |
Wallace P, Haernvall K, Ribitsch D, et al. (2017) PpEst is a novel PBAT degrading polyesterase identified by proteomic screening of Pseudomonas pseudoalcaligenes. Appl Microbiol Biotechnol 101: 2291-2303. https://doi.org/10.1007/s00253-016-7992-8
|
| [80] |
Martínez-Tobón D, Gul M, Elias A, et al. (2018) Polyhydroxybutyrate (PHB) biodegradation using bacterial strains with demonstrated and predicted PHB depolymerase activity. Appl Microbiol Biotechnol 102: 8049-8067. https://doi.org/10.1007/s00253-018-9153-8
|
| [81] |
Liu X, Hille P, Zheng M, et al. (2019) Diversity of polyester degrading bacteria in surface sediments from Yangtze River Estuary. AIP Conf Proc 2122: 020063. https://doi.org/10.1063/1.5116502
|
| [82] |
Qiu L, Yin X, Liu T, et al. (2020) Biodegradation of bis (2-hydroxyethyl) terephthalate by a newly isolated Enterobacter sp. HY1 and characterization of its esterase properties. J Basic Microbiol 60: 699-711. https://doi.org/10.1002/jobm.202000053
|
| [83] |
Gaino E, Pronzato R (1989) Ultrastructural evidence of bacterial damage to Spongia officinalis fibres (Porifera, Demospongiae). Dis Aquat Organ 6: 67-74. https://www.int-res.com/articles/dao/6/d006p067.pdf
|
| [84] |
Reisser J, Shaw J, Hallegraeff G, et al. (2014) Millimeter-sized marine plastics: a new pelagic habitat for microorganisms and invertebrates. PloS One 9: e100289. https://doi.org/10.1371/journal.pone.0100289
|
| [85] |
Bryant J, Clemente T, Viviani D, et al. (2016) Diversity and activity of communities inhabiting plastic debris in the North Pacific Gyre. mSystems 1: 024-16. https://doi.org/10.1128/mSystems.00024-16
|
| [86] |
Kiessling T, Gutow L, Thiel M (2015) Marine litter as habitat and dispersal vector. Marine anthropogenic litter . Cham: Springer Open Elsevier 141-181. https://doi.org/10.1007/978-3-319-16510-3_6
|
| [87] |
Zelezniak A, Andrejev S, Ponomarova O, et al. (2015) Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci 112: 6449-6454. https://doi.org/10.1073/pnas.1421834112
|
| [88] |
Borchert E, Hammerschmidt K, Hentschel U, et al. (2021) Enhancing microbial pollutant degradation by integrating eco-evolutionary principles with environmental biotechnology. Trends Microbiol 29: 908-918. https://doi.org/10.1016/j.tim.2021.03.002
|
| [89] |
Tournier V, Topham C, Gilles A, et al. (2020) An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580: 216-219. https://doi.org/10.1038/s41586-020-2149-4
|
| [90] |
Lenfant N, Hotelier T, Bourne Y, et al. (2013) Proteins with an alpha/beta hydrolase fold: relationships between subfamilies in an ever-growing superfamily. Chem Biol Interact 203: 266-268. https://doi.org/10.1016/j.cbi.2012.09.003
|
| [91] |
Gricajeva A, Nadda A, Gudiukaite R (2021) Insights into polyester plastic biodegradation by carboxyl ester hydrolases. J Chem Technol Biotechnol 97: 359-380. https://doi.org/10.1002/jctb.6745
|
 microbiol-09-03-027-s001.pdf microbiol-09-03-027-s001.pdf |
 |






Figures(7) / Tables(2)

Denisse Galarza–Verkovitch, Onur Turak, Jutta Wiese, Tanja Rahn, Ute Hentschel, Erik Borchert. Bioprospecting for polyesterase activity relevant for PET degradation in marine Enterobacterales isolates[J]. AIMS Microbiology, 2023, 9(3): 518-539. doi: 10.3934/microbiol.2023027







 DownLoad:
DownLoad: