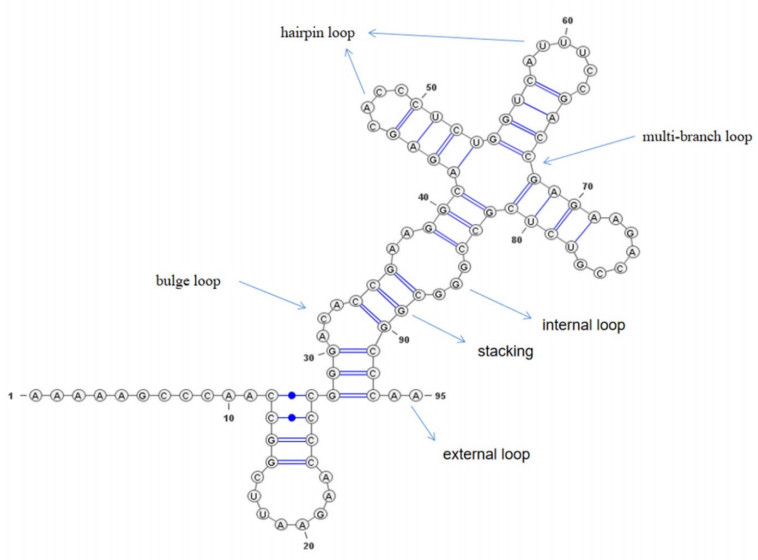
The RNA secondary structure is like a blueprint that holds the key to unlocking the mysteries of RNA function and 3D structure. It serves as a crucial foundation for investigating the complex world of RNA, making it an indispensable component of research in this exciting field. However, pseudoknots cannot be accurately predicted by conventional prediction methods based on free energy minimization, which results in a performance bottleneck. To this end, we propose a deep learning-based method called TransUFold to train directly on RNA data annotated with structure information. It employs an encoder-decoder network architecture, named Vision Transformer, to extract long-range interactions in RNA sequences and utilizes convolutions with lateral connections to supplement short-range interactions. Then, a post-processing program is designed to constrain the model's output to produce realistic and effective RNA secondary structures, including pseudoknots. After training TransUFold on benchmark datasets, we outperform other methods in test data on the same family. Additionally, we achieve better results on longer sequences up to 1600 nt, demonstrating the outstanding performance of Vision Transformer in extracting long-range interactions in RNA sequences. Finally, our analysis indicates that TransUFold produces effective pseudoknot structures in long sequences. As more high-quality RNA structures become available, deep learning-based prediction methods like Vision Transformer can exhibit better performance.
Citation: Yunxiang Wang, Hong Zhang, Zhenchao Xu, Shouhua Zhang, Rui Guo. TransUFold: Unlocking the structural complexity of short and long RNA with pseudoknots[J]. Mathematical Biosciences and Engineering, 2023, 20(11): 19320-19340. doi: 10.3934/mbe.2023854
The RNA secondary structure is like a blueprint that holds the key to unlocking the mysteries of RNA function and 3D structure. It serves as a crucial foundation for investigating the complex world of RNA, making it an indispensable component of research in this exciting field. However, pseudoknots cannot be accurately predicted by conventional prediction methods based on free energy minimization, which results in a performance bottleneck. To this end, we propose a deep learning-based method called TransUFold to train directly on RNA data annotated with structure information. It employs an encoder-decoder network architecture, named Vision Transformer, to extract long-range interactions in RNA sequences and utilizes convolutions with lateral connections to supplement short-range interactions. Then, a post-processing program is designed to constrain the model's output to produce realistic and effective RNA secondary structures, including pseudoknots. After training TransUFold on benchmark datasets, we outperform other methods in test data on the same family. Additionally, we achieve better results on longer sequences up to 1600 nt, demonstrating the outstanding performance of Vision Transformer in extracting long-range interactions in RNA sequences. Finally, our analysis indicates that TransUFold produces effective pseudoknot structures in long sequences. As more high-quality RNA structures become available, deep learning-based prediction methods like Vision Transformer can exhibit better performance.

| [1] |
J. A. Shapiro, Revisiting the central dogma in the 21st century, Ann. N. Y. Acad. Sci., 1178 (2009), 6-28. https://doi.org/10.1111/j.1749-6632.2009.04990.x doi: 10.1111/j.1749-6632.2009.04990.x

|
| [2] |
T. A. Lincoln, G. F. Joyce, Self-sustained replication of an RNA enzyme, Science, 323 (2009), 1229-1232. https://doi.org/10.1126/science.1167856 doi: 10.1126/science.1167856
|
| [3] |
P. V. Ryder, D. A. Lerit, RNA localization regulates diverse and dynamic cellular processes, Traffic, 19 (2018), 496-502. https://doi.org/10.1111/tra.12571 doi: 10.1111/tra.12571
|
| [4] | E. Westhof, P. Auffinger, RNA tertiary structure, in Encyclopedia of Analytical Chemistry, (2000), 5222-5232. https://doi.org/10.1002/9780470027318.a1428 |
| [5] |
F. E. Reyes, C. R. Schwartz, J. A. Tainer, R. P. Rambo, Methods for using new conceptual tools and parameters to assess RNA structure by small-angle X-ray scattering, Methods Enzymol., 549 (2014), 235-263. https://doi.org/10.1016/B978-0-12-801122-5.00011-8 doi: 10.1016/B978-0-12-801122-5.00011-8
|
| [6] |
C. Helmling, S. Keyhani, F. Sochor, B. Fürtig, M. Hengesbach, H. Schwalbe, Rapid NMR screening of RNA secondary structure and binding, J. Biomol. NMR, 63 (2015), 67-76. https://doi.org/10.1007/s10858-015-9967-y doi: 10.1007/s10858-015-9967-y
|
| [7] |
R. Stark, M. Grzelak, J. Hadfield, RNA sequencing: the teenage years, Nat. Rev. Genet., 20 (2019), 631-656. https://doi.org/10.1038/s41576-019-0150-2 doi: 10.1038/s41576-019-0150-2
|
| [8] |
M. Zuker, P. Stiegler, Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information, Nucleic Acids Res., 9 (1981), 133-148. https://doi.org/10.1093/nar/9.1.133 doi: 10.1093/nar/9.1.133
|
| [9] |
D. H. Turner, D. H. Mathews, NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure, Nucleic Acids Res., 38 (2010), D280-D282. https://doi.org/10.1093/nar/gkp892 doi: 10.1093/nar/gkp892
|
| [10] |
M. Zuker, Mfold web server for nucleic acid folding and hybridization prediction, Nucleic Acids Res., 31 (2003), 3406-3415. https://doi.org/10.1093/nar/gkg595 doi: 10.1093/nar/gkg595
|
| [11] | N. R. Markham, M. Zuker, UNAFold: software for nucleic acid folding and hybridization, in Bioinformatics, 453 (2008), 3-31. https://doi.org/10.1007/978-1-60327-429-6_1 |
| [12] |
I. L. Hofacker, W. Fontana, P. F. Stadler, L. S. Bonhoeffer, M. Tacker, P. Schuster, Fast folding and comparison of RNA secondary structures, Monatsh. Chem. Mon., 125 (1994), 167-188. https://doi.org/10.1007/BF00818163 doi: 10.1007/BF00818163
|
| [13] |
S. Bellaousov, J. S. Reuter, M. G. Seetin, D. H. Mathews, RNAstructure: web servers for RNA secondary structure prediction and analysis, Nucleic Acids Res., 41 (2013), W471-W474. https://doi.org/10.1093/nar/gkt290 doi: 10.1093/nar/gkt290
|
| [14] |
L. Huang, H. Zhang, D. Deng, K. Zhao, K. Liu, D. A. Hendrix, et al., LinearFold: linear-time approximate RNA folding by 5'-to-3'dynamic programming and beam search, Bioinformatics, 35 (2019), i295-i304. https://doi.org/10.1093/bioinformatics/btz375 doi: 10.1093/bioinformatics/btz375
|
| [15] |
X. Wang, J. Tian, Dynamic programming for NP-hard problems, Procedia Eng., 15 (2011), 3396-3400. https://doi.org/10.1016/j.proeng.2011.08.636 doi: 10.1016/j.proeng.2011.08.636
|
| [16] |
E. Rivas, S. R. Eddy, A dynamic programming algorithm for RNA structure prediction including pseudoknots, J. Mol. Biol., 285 (1999), 2053-2068. https://doi.org/10.1006/jmbi.1998.2436 doi: 10.1006/jmbi.1998.2436
|
| [17] |
R. M. Dirks, N. A. Pierce, A partition function algorithm for nucleic acid secondary structure including pseudoknots, J. Comput. Chem., 24 (2003), 1664-1677. https://doi.org/10.1002/jcc.10296 doi: 10.1002/jcc.10296
|
| [18] |
X. Xu, P. Zhao, S. J. Chen, Vfold: a web server for RNA structure and folding thermodynamics prediction, PloS One, 9 (2014), e107504. https://doi.org/10.1371/journal.pone.0107504 doi: 10.1371/journal.pone.0107504
|
| [19] | K. Sato, M. Hamada, Recent trends in RNA informatics: a review of machine learning and deep learning for RNA secondary structure prediction and RNA drug discovery, Briefings Bioinf., 24 (2023). https://doi.org/10.1093/bib/bbad186 |
| [20] |
T. Gong, F. Ju, D. Bu, Accurate prediction of RNA secondary structure including pseudoknots through solving minimum-cost flow with learned potentials, bioRxiv, (2022). https://doi.org/10.1101/2022.09.19.508461 doi: 10.1101/2022.09.19.508461
|
| [21] |
J. Ren, B. Rastegari, A. Condon, H. H. Hoos, HotKnots: heuristic prediction of RNA secondary structures including pseudoknots, RNA, 11 (2005), 1494-1504. https://doi.org/10.1261/rna.7284905 doi: 10.1261/rna.7284905
|
| [22] |
K. Sato, Y. Kato, M. Hamada, T. Akutsu, K. Asai, IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming, Bioinformatics, 27 (2011), i85-i93. https://doi.org/10.1093/bioinformatics/btr215 doi: 10.1093/bioinformatics/btr215
|
| [23] |
C. B. Do, D. A. Woods, S. Batzoglou, CONTRAfold: RNA secondary structure prediction without physics-based models, Bioinformatics, 22 (2006), e90-e98. https://doi.org/10.1093/bioinformatics/btl246 doi: 10.1093/bioinformatics/btl246
|
| [24] | S. Zakov, Y. Goldberg, M. Elhadad, M. Ziv-Ukelson, Rich parameterization improves RNA structure prediction, in Research in Computational Molecular Biology, 18 (2011), 1525-1542. https://doi.org/10.1007/978-3-642-20036-6_48 |
| [25] |
M. Akiyama, K. Sato, Y. Sakakibara, A max-margin training of RNA secondary structure prediction integrated with the thermodynamic model, J. Bioinf. Comput. Biol., 16 (2018), 1840025. https://doi.org/10.1142/S0219720018400255 doi: 10.1142/S0219720018400255
|
| [26] |
K. Sato, M. Akiyama, Y. Sakakibara, RNA secondary structure prediction using deep learning with thermodynamic integration, Nat. Commun., 12 (2021), 941. https://doi.org/10.1038/s41467-021-21194-4 doi: 10.1038/s41467-021-21194-4
|
| [27] |
H. Zhang, C. Zhang, Z. Li, C. Li, X. Wei, B. Zhang, et al., A new method of RNA secondary structure prediction based on convolutional neural network and dynamic programming, Front. Genet., 10 (2019), 467. https://doi.org/10.3389/fgene.2019.00467 doi: 10.3389/fgene.2019.00467
|
| [28] |
J. Singh, J. Hanson, K. Paliwal, Y. Zhou, RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning, Nat. Commun., 10 (2019), 5407. https://doi.org/10.1038/s41467-019-13395-9 doi: 10.1038/s41467-019-13395-9
|
| [29] | K. He, X. Zhang, S. Ren, J. Sun, Deep residual learning for image recognition, in 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), (2016), 770-778. https://doi.org/10.1109/CVPR.2016.90 |
| [30] | Z. Huang, W. Xu, K. Yu, Bidirectional LSTM-CRF models for sequence tagging, preprint, arXiv: 1508.01991. |
| [31] | X. Chen, Y. Li, R. Umarov, X. Gao, L. Song, RNA secondary structure prediction by learning unrolled algorithms, preprint, arXiv: 2002.05810. |
| [32] | A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, et al., Attention is all you need, preprint, arXiv: 1706.03762. |
| [33] |
L. Fu, Y. Cao, J. Wu, Q. Peng, Q. Nie, X. Xie, et al., UFold: fast and accurate RNA secondary structure prediction with deep learning, Nucleic Acids Res., 50 (2022), e14. https://doi.org/10.1093/nar/gkab1074 doi: 10.1093/nar/gkab1074
|
| [34] |
K. Darty, A. Denise, Y. Ponty, VARNA: interactive drawing and editing of the RNA secondary structure, Bioinformatics, 25 (2009), 1974. https://doi.org/10.1093/bioinformatics/btp250 doi: 10.1093/bioinformatics/btp250
|
| [35] | A. Dosovitskiy, L. Beyer, A. Kolesnikov, D. Weissenborn, X. Zhai, T. Unterthiner, et al., An image is worth 16x16 words: transformers for image recognition at scale, preprint, arXiv: 2010.11929. |
| [36] |
Z. Tan, Y. Fu, G. Sharma, D. H. Mathews, TurboFold Ⅱ: RNA structural alignment and secondary structure prediction informed by multiple homologs, Nucleic Acids Res., 45 (2017), 11570-11581. https://doi.org/10.1093/nar/gkx815 doi: 10.1093/nar/gkx815
|
| [37] |
M. F. Sloma, D. H. Mathews, Exact calculation of loop formation probability identifies folding motifs in RNA secondary structures, RNA, 22 (2016), 1808-1818. https://doi.org/10.1261/rna.053694.115 doi: 10.1261/rna.053694.115
|
| [38] |
I. Kalvari, E. P. Nawrocki, N. Ontiveros-Palacios, J. Argasinska, K. Lamkiewicz, M. Marz, et al., Rfam 14: expanded coverage of metagenomic, viral and microRNA families, Nucleic Acids Res., 49 (2021), D192-D200. https://doi.org/10.1093/nar/gkaa1047 doi: 10.1093/nar/gkaa1047
|
| [39] |
Y. Wang, Y. Liu, S. Wang, Z. Liu, Y. Gao, H. Zhang, et al., ATTfold: RNA secondary structure prediction with pseudoknots based on attention mechanism, Front. Genet., 11 (2020), 612086. https://doi.org/10.3389/fgene.2020.612086 doi: 10.3389/fgene.2020.612086
|
| [40] |
J. D. Watson, F. H. C. Crick, Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid, Nature, 171 (1953), 737-738. https://doi.org/10.1038/171737a0 doi: 10.1038/171737a0
|
| [41] |
G. Varani, W. H. McClain, The G·U wobble base pair, EMBO Rep., 1 (2000), 18-23. https://doi.org/10.1093/embo-reports/kvd001 doi: 10.1093/embo-reports/kvd001
|
| [42] |
E. J. Strobel, A. M. Yu, J. B. Lucks, High-throughput determination of RNA structures, Nat. Rev. Genet., 19 (2018), 615-634. https://doi.org/10.1038/s41576-018-0034-x doi: 10.1038/s41576-018-0034-x
|
| [43] |
S. Lusvarghi, J. Sztuba-Solinska, K. J. Purzycka, J. W. Rausch, S. F. J. Le Grice, RNA secondary structure prediction using high-throughput SHAPE, Biology, 2013 (2013), e50243. https://doi.org/10.3791/50243-v doi: 10.3791/50243-v
|











Figures(12) / Tables(7)

Yunxiang Wang, Hong Zhang, Zhenchao Xu, Shouhua Zhang, Rui Guo. TransUFold: Unlocking the structural complexity of short and long RNA with pseudoknots[J]. Mathematical Biosciences and Engineering, 2023, 20(11): 19320-19340. doi: 10.3934/mbe.2023854







 DownLoad:
DownLoad: