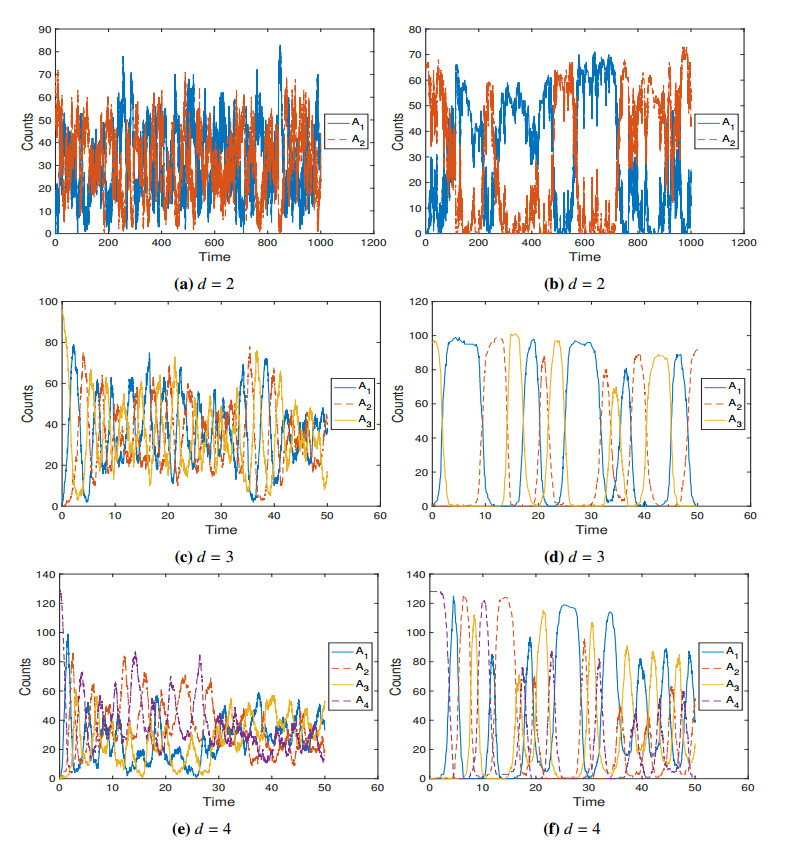
The Togashi Kaneko model (TK model) is a simple stochastic reaction network that displays discreteness-induced transitions between meta-stable patterns. Here we study a constrained Langevin approximation (CLA) of this model. This CLA, derived under the classical scaling, is an obliquely reflected diffusion process on the positive orthant and hence respects the constraint that chemical concentrations are never negative. We show that the CLA is a Feller process, is positive Harris recurrent and converges exponentially fast to the unique stationary distribution. We also characterize the stationary distribution and show that it has finite moments. In addition, we simulate both the TK model and its CLA in various dimensions. For example, we describe how the TK model switches between meta-stable patterns in dimension six. Our simulations suggest that, when the volume of the vessel in which all of the reactions that take place is large, the CLA is a good approximation of the TK model in terms of both the stationary distribution and the transition times between patterns.
Citation: Wai-Tong (Louis) Fan, Yifan (Johnny) Yang, Chaojie Yuan. Constrained Langevin approximation for the Togashi-Kaneko model of autocatalytic reactions[J]. Mathematical Biosciences and Engineering, 2023, 20(3): 4322-4352. doi: 10.3934/mbe.2023201
The Togashi Kaneko model (TK model) is a simple stochastic reaction network that displays discreteness-induced transitions between meta-stable patterns. Here we study a constrained Langevin approximation (CLA) of this model. This CLA, derived under the classical scaling, is an obliquely reflected diffusion process on the positive orthant and hence respects the constraint that chemical concentrations are never negative. We show that the CLA is a Feller process, is positive Harris recurrent and converges exponentially fast to the unique stationary distribution. We also characterize the stationary distribution and show that it has finite moments. In addition, we simulate both the TK model and its CLA in various dimensions. For example, we describe how the TK model switches between meta-stable patterns in dimension six. Our simulations suggest that, when the volume of the vessel in which all of the reactions that take place is large, the CLA is a good approximation of the TK model in terms of both the stationary distribution and the transition times between patterns.

| [1] |
Y. Togashi, K. Kaneko, Transitions induced by the discreteness of molecules in a small autocatalytic system, Phys. Rev. Lett., 86 (2001), 2459. https://doi.org/10.1103/PhysRevLett.86.2459 doi: 10.1103/PhysRevLett.86.2459

|
| [2] |
E. Bibbona, J. Kim, C. Wiuf, Stationary distributions of systems with discreteness-induced transitions, J. R. Soc. Interface, 17 (2020), 20200243. https://doi.org/10.1098/rsif.2020.0243 doi: 10.1098/rsif.2020.0243
|
| [3] |
M. Samoilov, S. Plyasunov, A. P. Arkin, Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations, Proc. Natl. Acad. Sci., 102 (2005), 2310–2315. https://doi.org/10.1073/pnas.0406841102 doi: 10.1073/pnas.0406841102
|
| [4] |
A. Awazu, K. Kaneko, Discreteness-induced transition in catalytic reaction networks, Phys. Rev. E, 76 (2007), 041915. https://doi.org/10.1103/PhysRevE.76.041915 doi: 10.1103/PhysRevE.76.041915
|
| [5] |
T. J. Kobayashi, Connection between noise-induced symmetry breaking and an information-decoding function for intracellular networks, Phys. Rev. Lett., 106 (2011), 228101. https://doi.org/10.1103/PhysRevLett.106.228101 doi: 10.1103/PhysRevLett.106.228101
|
| [6] |
T. Biancalani, T. Rogers, A. J. McKane, Noise-induced metastability in biochemical networks, Phys. Rev. E, 86 (2012), 010106. https://doi.org/10.1103/PhysRevE.86.010106 doi: 10.1103/PhysRevE.86.010106
|
| [7] |
Y. Togashi, K. Kaneko, Molecular discreteness in reaction-diffusion systems yields steady states not seen in the continuum limit, Phys. Rev. E, 70 (2004), 020901. https://doi.org/10.1103/PhysRevE.70.020901 doi: 10.1103/PhysRevE.70.020901
|
| [8] |
T. Butler, N. Goldenfeld, Fluctuation-driven turing patterns, Phys. Rev. E, 84 (2011), 011112. https://doi.org/10.1103/PhysRevE.84.011112 doi: 10.1103/PhysRevE.84.011112
|
| [9] |
T. To, N. Maheshri, Noise can induce bimodality in positive transcriptional feedback loops without bistability, Science, 327 (2010), 1142–1145. https://doi.org/10.1126/science.1178962 doi: 10.1126/science.1178962
|
| [10] |
R. Ma, J. Wang, Z. Hou, H. Liu, Small-number effects: a third stable state in a genetic bistable toggle switch, Phys. Rev. Lett., 109 (2012), 248107. https://doi.org/10.1103/PhysRevLett.109.248107 doi: 10.1103/PhysRevLett.109.248107
|
| [11] |
J. Sardanyés, T. Alarcón, Noise-induced bistability in the fate of cancer phenotypic quasispecies: a bit-strings approach, Sci. Rep., 8 (2018), 1027. https://doi.org/10.1038/s41598-018-19552-2 doi: 10.1038/s41598-018-19552-2
|
| [12] |
J. Sardanyés, A. Arderiu, S. F. Elena, T. Alarcón, Noise-induced bistability in the quasi-neutral coexistence of viral rnas under different replication modes, J. R. Soc. Interface, 15 (2018), 20180129. https://doi.org/10.1098/rsif.2018.0129 doi: 10.1098/rsif.2018.0129
|
| [13] |
T. Biancalani, L. Dyson, A. J. McKane, Noise-induced bistable states and their mean switching time in foraging colonies, Phys. Rev. Lett., 112 (2014), 038101. https://doi.org/10.1103/PhysRevLett.112.038101 doi: 10.1103/PhysRevLett.112.038101
|
| [14] |
B. Houchmandzadeh, M. Vallade, Exact results for a noise-induced bistable system, Phys. Rev. E, 91 (2015), 022115. https://doi.org/10.1103/PhysRevE.91.022115 doi: 10.1103/PhysRevE.91.022115
|
| [15] |
N. Saito, K. Kaneko, Theoretical analysis of discreteness-induced transition in autocatalytic reaction dynamics, Phys. Rev. E, 91 (2015), 022707. https://doi.org/10.1103/PhysRevE.91.022707 doi: 10.1103/PhysRevE.91.022707
|
| [16] |
L. Hoessly, C. Mazza, Stationary distributions and condensation in autocatalytic reaction networks, SIAM J. Appl. Math., 79 (2019), 1173–1196. https://doi.org/10.1137/18M1220340 doi: 10.1137/18M1220340
|
| [17] |
J. K. McSweeney, L. Popovic, Stochastically-induced bistability in chemical reaction systems, Ann. Appl. Probab., 24 (2014), 1226–1268. https://doi.org/10.1214/13-AAP946 doi: 10.1214/13-AAP946
|
| [18] |
T. Plesa, R. Erban, H. G. Othmer, Noise-induced mixing and multimodality in reaction networks, Eur. J. Appl. Math., 30 (2019), 887–911. https://doi.org/10.1017/S0956792518000517 doi: 10.1017/S0956792518000517
|
| [19] |
M. A. Al-Radhawi, D. D. Vecchio, E. D. Sontag, Multi-modality in gene regulatory networks with slow promoter kinetics, PLoS Comput. Biol., 15 (2019), e1006784. https://doi.org/10.1371/journal.pcbi.1006784 doi: 10.1371/journal.pcbi.1006784
|
| [20] | D. F. Anderson, T. G. Kurtz, Continuous time markov chain models for chemical reaction networks, in Design and Analysis of Biomolecular Circuits, (2011), 3–42. https://doi.org/10.1007/978-1-4419-6766-4_1 |
| [21] | M. Chen, From Markov Chains to Non-Equilibrium Particle Systems, World Scientific, 2004. https://doi.org/10.1142/5513 |
| [22] |
H. Kang, T. G. Kurtz, L. Popovic, Central limit theorems and diffusion approximations for multiscale markov chain models, Ann. Appl. Probab., 24 (2014), 721–759. https://doi.org/10.1214/13-AAP934 doi: 10.1214/13-AAP934
|
| [23] |
D. F. Anderson, D. J. Higham, S. C. Leite, R. J. Williams, On constrained langevin equations and (bio) chemical reaction networks, Multiscale Model. Simul., 17 (2019), 1–30. https://doi.org/10.1137/18M1190999 doi: 10.1137/18M1190999
|
| [24] |
S. C. Leite, R. J. Williams, A constrained langevin approximation for chemical reaction networks, Ann. Appl. Probab., 29 (2019), 1541–1608. https://doi.org/10.1214/18-AAP1421 doi: 10.1214/18-AAP1421
|
| [25] |
J. M. Harrison, H. J. Landau, L. A. Shepp, The stationary distribution of reflected brownian motion in a planar region, Ann. Appl. Probab., 13 (1985), 744–757. https://doi.org/10.1214/aop/1176992906 doi: 10.1214/aop/1176992906
|
| [26] |
P. Dupuis, H. Ishii, Sdes with oblique reflection on nonsmooth domains. Ann. Appl. Probab., 21 (1993), 554–580. https://doi.org/10.1214/aop/1176989415 doi: 10.1214/aop/1176989415
|
| [27] |
S. P. Meyn, R. L. Tweedie, Stability of markovian processes Ⅲ: Foster–lyapunov criteria for continuous-time processes, Adv. Appl. Probab., 25 (2016), 518–548. https://doi.org/10.2307/1427522 doi: 10.2307/1427522
|
| [28] | L. Stettner, On the existence and uniqueness of invariant measure for continuous time markov processes, 1986. Available from: https://apps.dtic.mil/sti/pdfs/ADA174758.pdf. |
| [29] |
R. Atar, A. Budhiraja, P. Dupuis, On positive recurrence of constrained diffusion processes, Ann. Probab., 29 (2001), 979–1000. https://doi.org/10.1214/aop/1008956699 doi: 10.1214/aop/1008956699
|
| [30] |
A. Budhiraja, C. Lee, Long time asymptotics for constrained diffusions in polyhedral domains, Stochastic Processes Appl., 117 (2007), 1014–1036. https://doi.org/10.1016/j.spa.2006.11.007 doi: 10.1016/j.spa.2006.11.007
|
| [31] |
W. Kang, K. Ramanan, Characterization of stationary distributions of reflected diffusions, Ann. Appl. Probab., 24 (2014), 1329–1374. https://doi.org/10.1214/13-AAP947 doi: 10.1214/13-AAP947
|
| [32] |
J. M. Harrison, R. J. Williams, Brownian models of open queueing networks with homogeneous customer populations, Stochastics, 22 (1987), 77–115. https://doi.org/10.1080/17442508708833469 doi: 10.1080/17442508708833469
|
| [33] |
J. G. Dai, J. M. Harrison, Reflected brownian motion in an orthant: numerical methods for steady-state analysis, Ann. Appl. Probab., 2 (1992), 65–86. https://doi.org/10.1214/aoap/1177005771 doi: 10.1214/aoap/1177005771
|
| [34] |
D. T. Gillespie, Exact stochastic simulation of coupled chemical reactions, J. Phys. Chem., 81 (1977), 2340–2361. https://doi.org/10.1021/j100540a008 doi: 10.1021/j100540a008
|
| [35] |
M. Bossy, E. Gobet, D. Talay, A symmetrized euler scheme for an efficient approximation of reflected diffusions, J. Appl. Probab., 41 (2004), 877–889. https://doi.org/10.1239/jap/1091543431 doi: 10.1239/jap/1091543431
|
| [36] |
W. L. Fan, Discrete approximations to local times for reflected diffusions, Electron. Commun. Probab., 21 (2016), 1–12. https://doi.org/10.1214/16-ECP4694 doi: 10.1214/16-ECP4694
|
| [37] |
Z. Chen, W. L. Fan, Hydrodynamic limits and propagation of chaos for interacting random walks in domains, Ann. Appl. Probab., 27 (2017), 1299–1371. https://doi.org/10.1214/16-AAP1208 doi: 10.1214/16-AAP1208
|
| [38] | S. Karlin, H. E. Taylor, A Second Course in Stochastic Processes, Elsevier, 1981. |
| [39] |
R. T. Powers, E. Størmer, Free states of the canonical anticommutation relations, Commun. Math. Phys., 16 (1970), 1–33. https://doi.org/10.1007/BF01645492 doi: 10.1007/BF01645492
|
| [40] | S. S. Dragomir, M. City, Some gronwall type inequalities and applications, 2002. Available from: https://rgmia.org/papers/monographs/standard.pdf. |
| [41] |
E. B. Dynkin, A. A. Yushkevich, Strong markov processes, Theory Probab. Appl., 1 (1956), 134–139. https://doi.org/10.1137/1101012 doi: 10.1137/1101012
|
| [42] | Nils Berglund, Long-time dynamics of stochastic differential equations, preprint, arXiv: 2106.12998. |
| [43] |
A. Sarantsev, Reflected brownian motion in a convex polyhedral cone: tail estimates for the stationary distribution, J. Theor. Probab., 30 (2017), 1200–1223. https://doi.org/10.1007/s10959-016-0674-8 doi: 10.1007/s10959-016-0674-8
|
| [44] |
W. Kang, K. Ramanan, On the submartingale problem for reflected diffusions in domains with piecewise smooth boundaries, Ann. Probab., 45 (2017), 404–468. https://doi.org/10.1214/16-AOP1153 doi: 10.1214/16-AOP1153
|












Figures(13)

Wai-Tong (Louis) Fan, Yifan (Johnny) Yang, Chaojie Yuan. Constrained Langevin approximation for the Togashi-Kaneko model of autocatalytic reactions[J]. Mathematical Biosciences and Engineering, 2023, 20(3): 4322-4352. doi: 10.3934/mbe.2023201







 DownLoad:
DownLoad: