Citation: Shuwan Yin, Jia Zheng, Cangzhi Jia, Quan Zou, Zhengkui Lin, Hua Shi. UPFPSR: a ubiquitylation predictor for plant through combining sequence information and random forest[J]. Mathematical Biosciences and Engineering, 2022, 19(1): 775-791. doi: 10.3934/mbe.2022035

| [1] |
A. R. Farley, A. J. Link, Identification and quantification of protein posttranslational modifications, Methods Enzymol., 463 (2009), 725-763. doi: 10.1016/S0076-6879(09)63040-8. doi: 10.1016/S0076-6879(09)63040-8

|
| [2] | J. Jia, Y. Shen, W. Qiu, Identifying Lysine Succinylation Sites in Proteins by Broad Learning System and Optimizing Imbalanced Training Dataset via Randomly Labeling Samples, Wuhan Univ. J. Nat. Sci., (2021), 81-88. doi: 10.19823/j.cnki.1007-1202.2021.0005. |
| [3] |
C. Ou, H. Pi, C. Chien, Control of protein degradation by E3 ubiquitin ligases in Drosophila eye development, Trends Genet., 19 (2003), 382-389. doi: 10.1016/S0168-9525(03)00146-X. doi: 10.1016/S0168-9525(03)00146-X
|
| [4] |
J. Herrmann, L. Lerman, A. Lerman, Ubiquitin and Ubiquitin-Like Proteins in Protein Regulation, Circul. Res., 100 (2007), 1276-1291. doi: 10.1161/01.res.0000264500.11888.f0. doi: 10.1161/01.res.0000264500.11888.f0
|
| [5] |
R. Welchman, C. Gordon, R. Mayer, Ubiquitin and ubiquitin-like proteins as multifunctional signals, Nat. Rev. Mol. Cell Biol., 6 (2005), 599-609. doi: 10.1038/nrm1700. doi: 10.1038/nrm1700
|
| [6] |
Y. Tu, C. Chen, J. Pan, J. Xu, Z. Zhou, C. Wang, The Ubiquitin Proteasome Pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis, Int. J. Clin. Exp. Pathol., 5 (2012), 726-738. doi: 10.3109/15513815.2012.659410. doi: 10.3109/15513815.2012.659410
|
| [7] |
A. Schwartz, A. Ciechanover, The ubiquitin-proteasome pathway and pathogenesis of human diseases, Annu. Rev. Med., 50 (1999), 57-74. doi: 10.1146/annurev.med.50.1.57. doi: 10.1146/annurev.med.50.1.57
|
| [8] |
X. Chen, J. Qiu, S. Shi, S. Suo, S. Huang, R. Liang, Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites, Bioinformatics, 29 (2013), 1614-1622. doi: 10.1093/bioinformatics/btt196. doi: 10.1093/bioinformatics/btt196
|
| [9] |
W. Qiu, C. Xu, X. Xiao, D. Xu, Computational Prediction of Ubiquitination Proteins Using Evolutionary Profiles and Functional Domain Annotation, Curr. Genomics, 20 (2019), 389-399. doi: 10.2174/1389202919666191014091250. doi: 10.2174/1389202919666191014091250
|
| [10] |
C. Tung, S. Ho, Computational identification of ubiquitylation sites from protein sequences, BMC Bioinf., 9 (2008), 310. doi: 10.1186/1471-2105-9-310. doi: 10.1186/1471-2105-9-310
|
| [11] |
V. Nguyen, K. Huang, C. Huang, K. Lai, T. Lee, A new scheme to characterize and identify protein ubiquitination sites, IEEE/ACM Trans. Comput. Biol. Bioinf., 14 (2016), 393-403. doi: 10.1109/TCBB.2016.2520939. doi: 10.1109/TCBB.2016.2520939
|
| [12] |
C. Huang, M. Su, H. Kao, J. Jhong, S. Weng, T. Lee, UbiSite: incorporating two-layered machine learning method with substrate motifs to predict ubiquitin-conjugation site on lysines, BMC Syst. Biol., 10 (2016), 6. doi: 10.1186/s12918-015-0246-z. doi: 10.1186/s12918-015-0246-z
|
| [13] |
J. Chen, J. Zhao, S. Yang, Z. Chen, Z. Zhang, Prediction of protein ubiquitination sites in Arabidopsis thaliana, Curr. Bioinf., 14 (2019), 614-620. doi: 10.2174/1574893614666190311141647. doi: 10.2174/1574893614666190311141647
|
| [14] |
X. Cui, Z. Yu, B. Yu, M. Wang, B. Tian, Q. Ma, UbiSitePred: A novel method for improving the accuracy of ubiquitination sites prediction by using LASSO to select the optimal Chous pseudo components, Chemometr. Intellig. Lab. Syst., 184 (2019), 28-43. doi: 10.1016/j.chemolab.2018.11.012. doi: 10.1016/j.chemolab.2018.11.012
|
| [15] | M. Mosharaf, F. Ahmed, M. Hassan, S. Tasmia, M. Mollah, In Silico Prediction of Protein Ubiquitination Sites by Using Binary Encoding on Arabidopsis thaliana, Int. J. Statist. Sci., 18 (2019), 65-76. |
| [16] |
M. Mosharaf, M. Hassan, F. Ahmed, M. Khatun, M. Moni, M. Mollah, Computational prediction of protein ubiquitination sites mapping on Arabidopsis thaliana, Comput. Biol. Chem., 85 (2020), 107238. doi: 10.1016/j.compbiolchem.2020.107238. doi: 10.1016/j.compbiolchem.2020.107238
|
| [17] |
Z. Chen, Y. Zhou, J. Song, Z. Zhang, hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties, BBA-Proteins Proteomics, 1834 (2013), 1461-1467. doi: 10.1016/j.bbapap.2013.04.006. doi: 10.1016/j.bbapap.2013.04.006
|
| [18] |
W. Qiu, X. Xiao, W. Lin, K. Chou, iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model, J. Biomol. Struct. Dyn., 33 (2015), 1731-1742. doi: 10.1080/07391102.2014.968875. doi: 10.1080/07391102.2014.968875
|
| [19] |
B. Cai, X. Jiang, Computational methods for ubiquitination site prediction using physicochemical properties of protein sequences, BMC Bioinf., 17 (2016), 1-12. doi: 10.1186/s12859-016-0959-z. doi: 10.1186/s12859-016-0959-z
|
| [20] |
J. Wang, W. Huang, M. Tsai, K. Hsu, H. Huang, S. Ho, ESA-UbiSite: accurate prediction of human ubiquitination sites by identifying a set of effective negatives, Bioinformatics, 33 (2017), 661-668. doi: 10.1093/bioinformatics/btw701. doi: 10.1093/bioinformatics/btw701
|
| [21] |
F. He, R. Wang, J. Li, L. Bao, D. Xu, X. Zhao, Large-scale prediction of protein ubiquitination sites using a multimodal deep architecture, BMC Syst. Biol., 12 (2018), 81-90. doi: 10.1186/s12918-018-0628-0. doi: 10.1186/s12918-018-0628-0
|
| [22] | S. Yadav, M. Gupta, A. Bist, Prediction of ubiquitination sites using UbiNets, Adv. Fuzzy Syst., 2018 (2018). doi: 10.1155/2018/5125103. |
| [23] |
H. Fu, Y. Yang, X. Wang, H. Wang, Y. Xu, DeepUbi: a deep learning framework for prediction of ubiquitination sites in proteins, BMC Bioinf, , 20 (2019), 86. doi: 10.1186/s12859-019-2677-9. doi: 10.1186/s12859-019-2677-9
|
| [24] |
H. Wang, Z. Wang, Z. Li, T. Lee, Incorporating Deep Learning With Word Embedding to Identify Plant Ubiquitylation Sites, Front. Cell. Dev. Biol., 8 (2020), 572195. doi: 10.3389/fcell.2020.572195. doi: 10.3389/fcell.2020.572195
|
| [25] |
H. Xu, J. Zhou, S. Lin, W. Deng, Y. Xue, PLMD: An updated data resource of protein lysine modifications, J. Genet. Genomics, 44 (2017), 243-250. doi: 10.1016/j.jgg.2017.03.007. doi: 10.1016/j.jgg.2017.03.007
|
| [26] |
B. Li, L. Hu, S. Niu, Y. Cai, K. Chou, Predict and analyze S-nitrosylation modification sites with the mRMR and IFS approaches, J. Proteomics, 75 (2012), 1654-1665. doi: 10.1016/j.jprot.2011.12.003. doi: 10.1016/j.jprot.2011.12.003
|
| [27] |
Y. Zhu, C. Jia, F. Li, J. Song, Inspector: a lysine succinylation predictor based on edited nearest-neighbor undersampling and adaptive synthetic oversampling, Anal. Biochem., 593 (2020), 113592. doi: 10.1016/j.ab.2020.113592. doi: 10.1016/j.ab.2020.113592
|
| [28] |
C. Jia, Y. Zuo, Q. Zou, O-GlcNAcPRED-Ⅱ: an integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique, Bioinformatics, 34 (2018), 2029-2036. doi: 10.1093/bioinformatics/bty039. doi: 10.1093/bioinformatics/bty039
|
| [29] |
Z. Chen, P. Zhao, F. Li, L. André, T. Marquez-Lago, Y. Wang, et al., iFeature: a python package and web server for features extraction and selection from protein and peptide sequences, Bioinformatics, 34 (2018), 2499-2502. doi: 10.1093/bioinformatics/bty140. doi: 10.1093/bioinformatics/bty140
|
| [30] |
H. Shen, K. Chou, PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition, Anal. Biochem., 373 (2008), 386-388. doi: 10.1016/j.ab.2007.10.012. doi: 10.1016/j.ab.2007.10.012
|
| [31] |
T. Li, R. Song, Q. Yin, M. Gao, Y. Chen, Identification of S-nitrosylation sites based on multiple features combination, Sci. Rep., 9 (2019), 3098. doi: 10.1038/s41598-019-39743-9. doi: 10.1038/s41598-019-39743-9
|
| [32] | Q. Wuyun, W. Zheng, Y. Zhang, J. Ruan, G. Hu, Improved Species-Specific Lysine Acetylation Site Prediction Based on a Large Variety of Features Set, PLoS One, 11 (2016), e0155370. doi: 10.1371/journal.pone.0155370. |
| [33] |
W. Qiu, A. Xu, Z. Xu, C. Zhang, X. Xiao, Identifying Acetylation Protein by Fusing Its PseAAC and Functional Domain Annotation, Front. Bioeng. Biotechnol., 7 (2019), 311. doi: 10.3389/fbioe.2019.00311. doi: 10.3389/fbioe.2019.00311
|
| [34] |
Z. Chen, P. Zhao, F. Li, T. Marquez-Lago, A. Leier, J. Revote, et al., iLearn: an integrated platform and meta-learner for feature engineering, machine learning analysis and modeling of DNA, RNA and protein sequence data, Brief. Bioinf., 21 (2019), 1047-1057. doi: 10.1093/bib/bbz041. doi: 10.1093/bib/bbz041
|
| [35] | S. Shi, J. Qiu, X. Sun, S. Suo, S. Huang, R. Liang, PMeS: Prediction of Methylation Sites Based on Enhanced Feature Encoding Scheme, PLoS One, 7 (2012), e38772. doi: 10.1371/journal.pone.0038772. |
| [36] | L. Breiman, Random forests, Mach. Learn., 45 (2001), 5-32. doi: 10.1023/A:1010933404324. |
| [37] |
J. Jia, Z. Liu, X. Xiao, B. Liu, K. Chou, pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach, J. Theor. Biol., 394 (2016), 223-230. doi: 10.1016/j.jtbi.2016.01.020. doi: 10.1016/j.jtbi.2016.01.020
|
| [38] |
M. Hasan, S. Yang, Y. Zhou, M. Mollah, SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties, Mol. Biosyst., 12 (2016), 786-795. doi: 10.1039/C5MB00853K. doi: 10.1039/C5MB00853K
|
| [39] |
M. Hasan, M. Khatun, M. Mollah, C. Yong, D. Guo, A systematic identification of species-specific protein succinylation sites using joint element features information, Int. J. Nanomed., 12 (2017), 1-13. doi: 10.2147/IJN.S140875. doi: 10.2147/IJN.S140875
|
| [40] |
H. Ismail, A. Jones, J. Kim, R. Newman, D. Kc, RF-Phos: A Novel General Phosphorylation Site Prediction Tool Based on Random Forest, BioMed. Res. Int., 2016 (2016), 3281590. doi: 10.1155/2016/3281590. doi: 10.1155/2016/3281590
|
| [41] |
H. AL-barakati, H. Saigo, R. Newman, B. Dukka, RF-GlutarySite: a random forest based predictor for glutarylation sites, Mol. Omics, 15 (2019), 189-204. doi: 10.1039/C9MO00028C. doi: 10.1039/C9MO00028C
|
| [42] | V. Vapnik, A. Lerner, Recognition of patterns with help of generalized portraits, Avtomat. Telemekh., 24 (1963), 774-780. |
| [43] |
C. Jia, M. Zhang, C. Fan, F. Li, J. Song, Formator: predicting lysine formylation sites based on the most distant undersampling and safe-level synthetic minority oversampling, IEEE/ACM Trans. Comput. Biol. Bioinf., 18 (2019), 1937-1945. doi: 10.1109/TCBB.2019.2957758. doi: 10.1109/TCBB.2019.2957758
|
| [44] |
R. Wang, Z. Wang, H. Wang, Y. Pang, T. Lee, Characterization and identification of lysine crotonylation sites based on machine learning method on both plant and mammalian, Sci. Rep., 10 (2020), 20447. doi: 10.1038/s41598-020-77173-0. doi: 10.1038/s41598-020-77173-0
|
| [45] | S. Suo, J. Qiu, S. Shi, X. Sun, S. Huang, X. Chen, et al., Position-specific analysis and prediction for protein lysine acetylation based on multiple features, PLoS One, 7 (2012), e49108. doi: 10.1371/journal.pone.0049108. |
| [46] |
H. Al-Barakati, E. McConnell, L. Hicks, L. Poole, R. Newman, SVM-SulfoSite: a support vector machine based predictor for sulfenylation sites, Sci. Rep., 8 (2018), 1-9. doi: 10.1038/s41598-018-29126-x. doi: 10.1038/s41598-018-29126-x
|
| [47] |
J. Raikwal, K. Saxena, Performance Evaluation of SVM and K-Nearest Neighbor Algorithm over Medical Data set, Int. J. Comput. Appl., 50 (2012), 35-39. doi: 10.5120/7842-1055. doi: 10.5120/7842-1055
|
| [48] |
Q. Ning, Z. Ma, X. Zhao, dForml(KNN)-PseAAC: Detecting Formylation sites from protein sequences using K-nearest neighbor algorithm via Chous 5-step rule and Pseudo components, J. Theor. Biol., 470 (2019), 43-49. doi: 10.1016/j.jtbi.2019.03.011. doi: 10.1016/j.jtbi.2019.03.011
|
| [49] |
K. Mittal, G. Aggarwal, P. Mahajan, Performance study of K-nearest neighbor classifier and K-means clustering for predicting the diagnostic accuracy, Int. J. Inf. Technol., 11 (2019), 535-540. doi: 10.1007/s41870-018-0233-x. doi: 10.1007/s41870-018-0233-x
|
| [50] | A. Singh, N. Malka, R. Lakshmiganthan, Impact of Different Data Types on Classifier Performance of Random Forest, Naï ve Bayes, and K-Nearest Neighbors Algorithms, Int. J. Adv. Comput. Sci. Appl., 8 (2017), 1-10. doi: 10.14569/ijacsa.2017.081201. |
| [51] |
M. Khatun, M. Hasan, Prediction of protein Post-Translational Modification sites: An overview, Ann. Proteom. Bioinf., 2 (2018), 49-57. doi: 10.29328/journal.apb.1001005. doi: 10.29328/journal.apb.1001005
|
| [52] |
A. Zamir, H. Khan, T. Iqbal, N. Yousaf, F. Aslam, A.Anjum, et al., Phishing web site detection using diverse machine learning algorithms, Electron. Libr., 38 (2020), 65-80. doi: 10.1108/EL-05-2019-0118. doi: 10.1108/EL-05-2019-0118
|
| [53] |
Y. Pan, H. Gao, H. Lin, Z. Liu, L. Tang, S. Li, Identification of Bacteriophage Virion Proteins Using Multinomial Nave Bayes with g-Gap Feature Tree, Int. J. Mol. Sci., 19 (2018), 1779. doi: 10.3390/ijms19061779. doi: 10.3390/ijms19061779
|
| [54] | G. Webb, N. Bayes, Encyclopedia of Machine Learning, in Springer US (eds. C. Sammut and G. I. Webb), Academic Press, (2010), 613-624. doi: 10.1007/978-0-387-30164-8. |
| [55] |
F. Li, J. Chen, Z. Ge, Y. Wen, Y. Yue, M. Hayashida, et al., Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework, Brief. Bioinf., 22 (2020), 2126-2140. doi: 10.1093/bib/bbaa049. doi: 10.1093/bib/bbaa049
|
| [56] | R. Xie, J. Li, J. Wang, W. Dai, A. Leier, T. Marquez-Lago, et al., DeepVF: a deep learning-based hybrid framework for identifying virulence factors using the stacking strategy, Brief. Bioinf., 22 (2020). doi: 10.1093/bib/bbaa125. |
| [57] | M. Wu, S. Pan, L. Du, X. Zhu, Learning Graph Neural Networks with Positive and Unlabeled Nodes, preprint, arXiv: 2103.04683. |
| [58] |
A. Strokach, T. Lu, P. Kim, ELASPIC2 (EL2): Combining Contextualized Language Models and Graph Neural Networks to Predict Effects of Mutations, J. Mol. Biol., 433 (2021), 166810. doi: 10.1016/j.jmb.2021.166810. doi: 10.1016/j.jmb.2021.166810
|
| [59] |
L. Zhang, P. Yang, H. Feng, Q. Zhao, H. Liu, Using network distance analysis to predict lncRNA-miRNA interactions, Interdiscip. Sci., 13 (2021), 535-545. doi: 10.1007/s12539-021-00458-z. doi: 10.1007/s12539-021-00458-z
|
| [60] |
V. Vacic, L. Iakoucheva, P. Radivojac, Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments, Bioinformatics, 22 (2006), 1536-1537. doi: 10.1093/bioinformatics/btl151. doi: 10.1093/bioinformatics/btl151
|
 mbe-19-01-035-Supplementary.pdf mbe-19-01-035-Supplementary.pdf |
 |
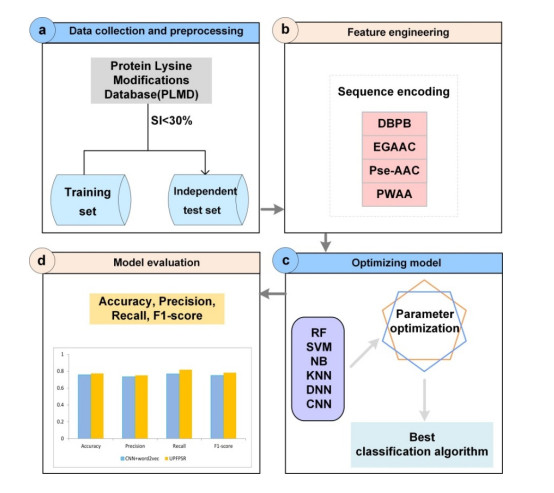
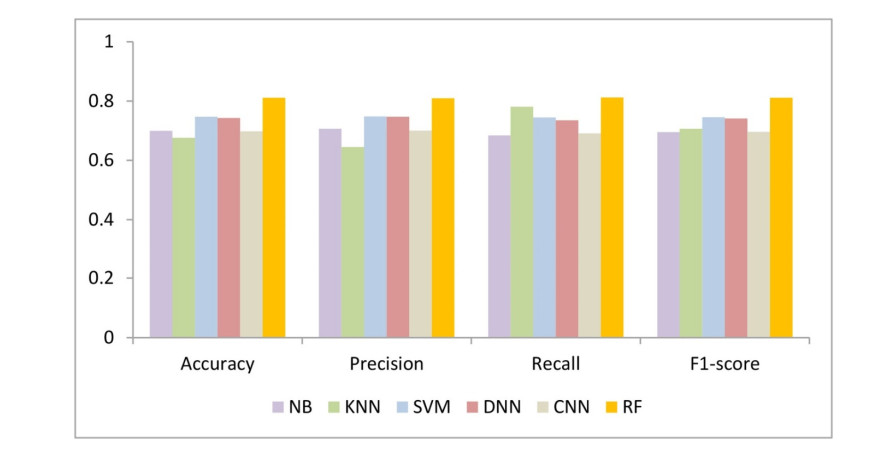
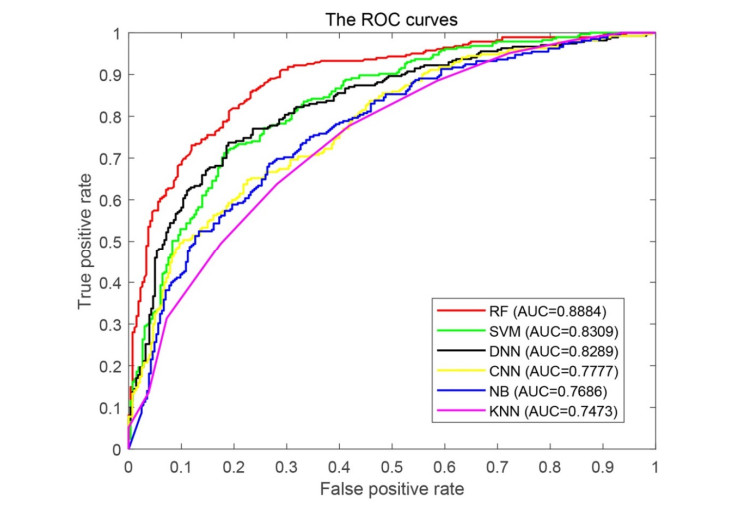
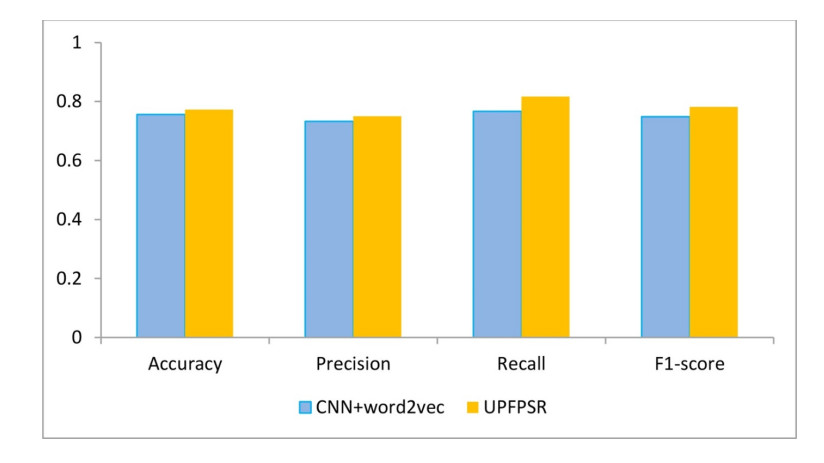
Figures(5) / Tables(2)

Shuwan Yin, Jia Zheng, Cangzhi Jia, Quan Zou, Zhengkui Lin, Hua Shi. UPFPSR: a ubiquitylation predictor for plant through combining sequence information and random forest[J]. Mathematical Biosciences and Engineering, 2022, 19(1): 775-791. doi: 10.3934/mbe.2022035







 DownLoad:
DownLoad: