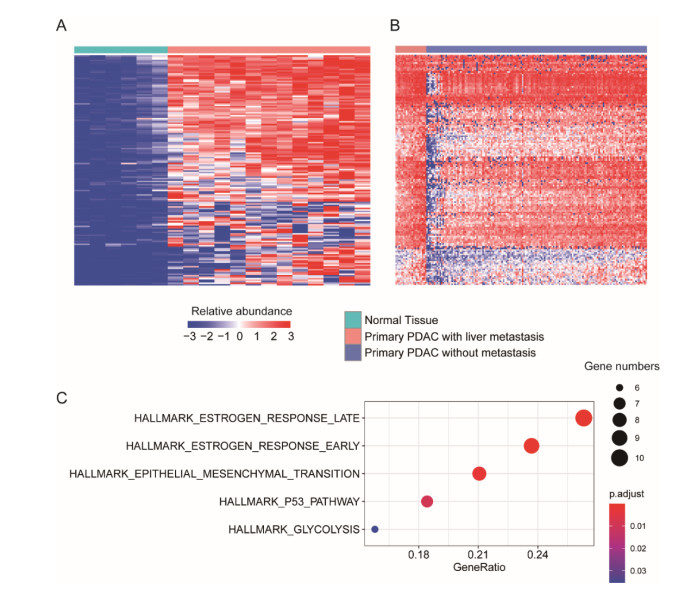
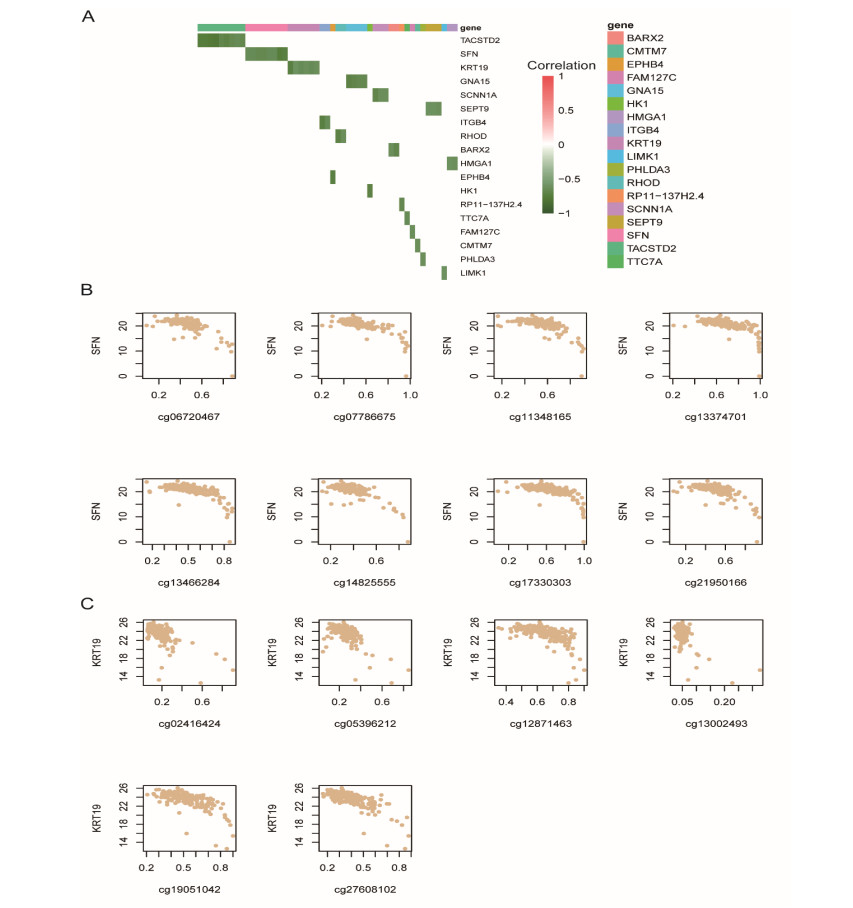
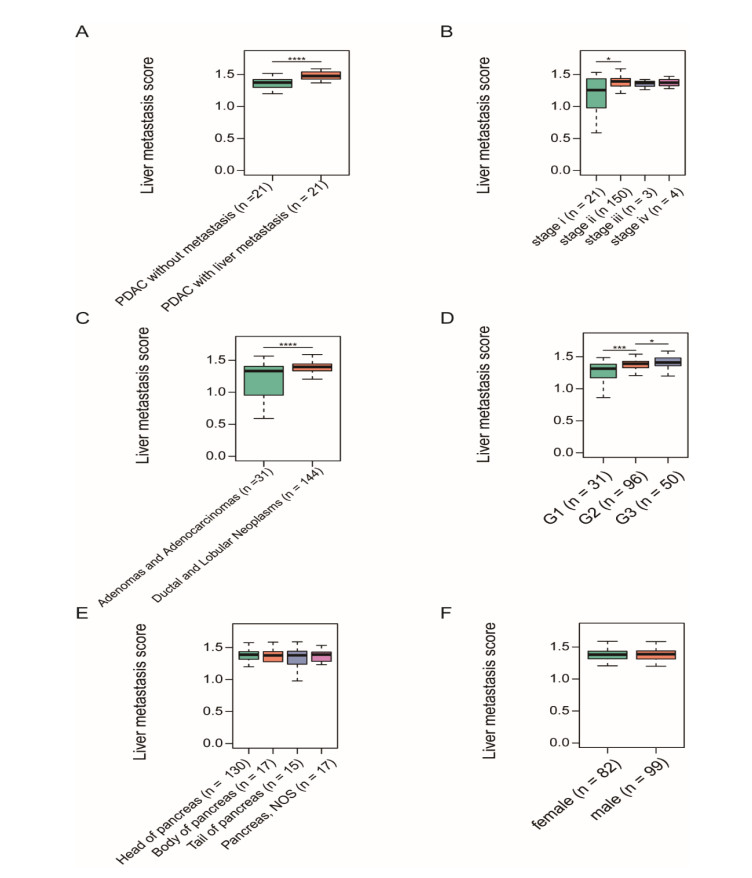
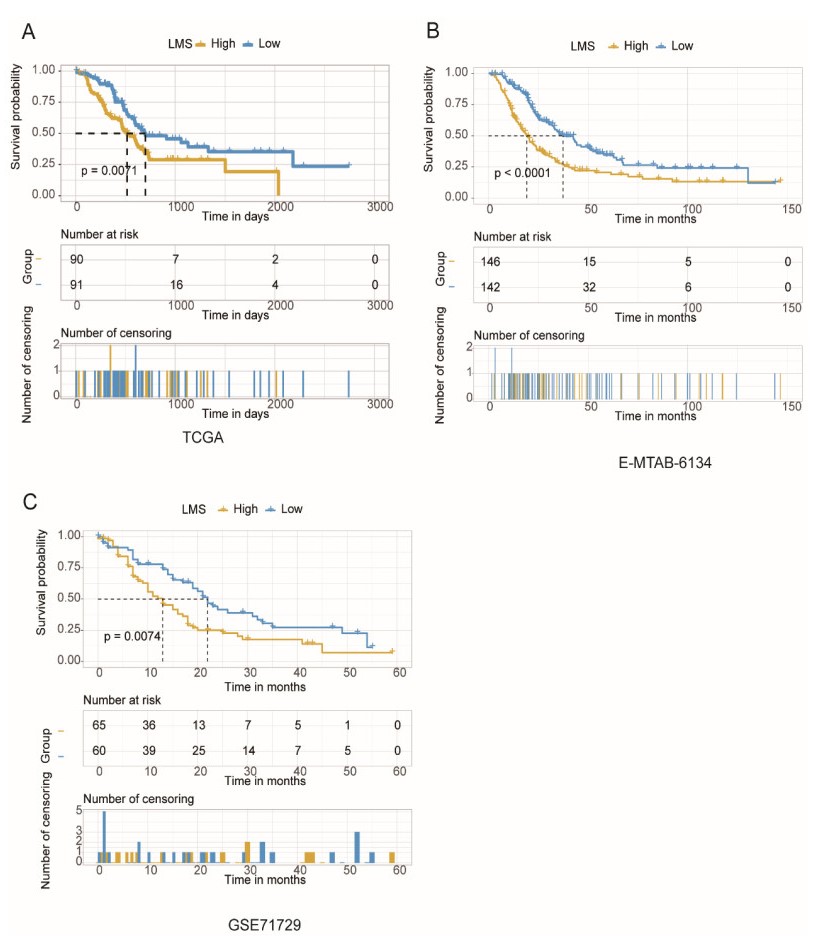
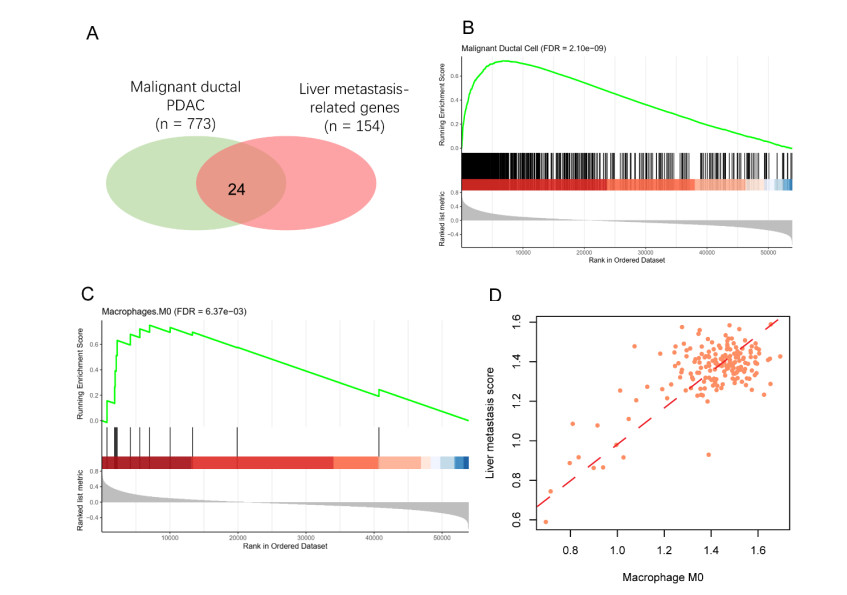
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies worldwide. However, the mechanisms underlying the acquisition of the metastatic potential in PDAC has not been well understood. In this study, we identified a total of 154 genes upregulated in primary tissues of PDAC with liver metastasis using the Genome Cancer Atlas (TCGA) and GSE151580 cohorts. The epithelial-mesenchymal transition and glycolysis were enriched by the liver metastasis-related genes, indicating that the liver metastasis-related genes might be functionally relevant to liver metastasis in PDAC. Moreover, we also found that the liver metastasis-related genes were primarily regulated at epigenetic level. Particularly, SFN, a cell cycle checkpoint protein, and KRT19, a marker gene for ductal cells, were predicted to be regulated by multiple methylation sites at the promoter. Clinically, we for the first time defined a liver metastasis score (LMS), which was derived from liver metastasis-related genes, and closely associated with clinical characteristics such as disease type and tumor grade, in PDAC. Furthermore, we also divided the samples into high and low LMS groups using three cohorts with long-term follow-up (TCGA, GSE71729, and E-MTAB-6134), which exhibited significantly different prognostic outcomes across three PDAC cohorts, suggesting that the LMS might be a good indicator for risk stratification in PDAC. Furthermore, we also found that the liver metastasis-related genes were primarily expressed in malignant ductal cells by integrative analysis of the bulk and single-cell gene expression data. Moreover, the malignant ductal cells and M0 macrophages were highly correlated with LMS, indicating that the two cell types might function as tumor-promoting cells in PDAC. In summary, the systematic analysis for the first time defined an LMS score to evaluate the risk of liver metastasis in PDAC and revealed that malignant ductal cells might promote PDAC liver metastasis, which greatly improves our understanding of the liver metastasis-related genes, their underlying mechanisms, and association with prognosis in PDAC.
Citation: Yang Yu, Zhe Wang, Dai hai Mo, Zhen Wang, Gang Li. Transcriptome profiling reveals liver metastasis-associated genes in pancreatic ductal adenocarcinoma[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1708-1721. doi: 10.3934/mbe.2021088
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies worldwide. However, the mechanisms underlying the acquisition of the metastatic potential in PDAC has not been well understood. In this study, we identified a total of 154 genes upregulated in primary tissues of PDAC with liver metastasis using the Genome Cancer Atlas (TCGA) and GSE151580 cohorts. The epithelial-mesenchymal transition and glycolysis were enriched by the liver metastasis-related genes, indicating that the liver metastasis-related genes might be functionally relevant to liver metastasis in PDAC. Moreover, we also found that the liver metastasis-related genes were primarily regulated at epigenetic level. Particularly, SFN, a cell cycle checkpoint protein, and KRT19, a marker gene for ductal cells, were predicted to be regulated by multiple methylation sites at the promoter. Clinically, we for the first time defined a liver metastasis score (LMS), which was derived from liver metastasis-related genes, and closely associated with clinical characteristics such as disease type and tumor grade, in PDAC. Furthermore, we also divided the samples into high and low LMS groups using three cohorts with long-term follow-up (TCGA, GSE71729, and E-MTAB-6134), which exhibited significantly different prognostic outcomes across three PDAC cohorts, suggesting that the LMS might be a good indicator for risk stratification in PDAC. Furthermore, we also found that the liver metastasis-related genes were primarily expressed in malignant ductal cells by integrative analysis of the bulk and single-cell gene expression data. Moreover, the malignant ductal cells and M0 macrophages were highly correlated with LMS, indicating that the two cell types might function as tumor-promoting cells in PDAC. In summary, the systematic analysis for the first time defined an LMS score to evaluate the risk of liver metastasis in PDAC and revealed that malignant ductal cells might promote PDAC liver metastasis, which greatly improves our understanding of the liver metastasis-related genes, their underlying mechanisms, and association with prognosis in PDAC.

| [1] |
R. L. Siegel, K. D. Miller, A. Jemal, Cancer statistics, 2020, CA Cancer J. Clin., 70 (2020), 7–30. doi: 10.3322/caac.21590

|
| [2] |
E. Giovannetti, C. L. van der Borden, A. E. Frampton, A. Ali, O. Firuzi, G. J. Peters, Never let it go: Stopping key mechanisms underlying metastasis to fight pancreatic cancer, Semin. Cancer Biol., 44 (2017), 43–59. doi: 10.1016/j.semcancer.2017.04.006
|
| [3] |
C. Torres, P. J. Grippo, Pancreatic cancer subtypes: a roadmap for precision medicine, Ann. Med., 50 (2018), 277–287. doi: 10.1080/07853890.2018.1453168
|
| [4] |
H. Oweira, U. Petrausch, D. Helbling, J. Schmidt, M. Mannhart, A. Mehrabi, et al., Prognostic value of site-specific metastases in pancreatic adenocarcinoma: A Surveillance Epidemiology and End Results database analysis, World J. Gastroenterol., 23 (2017), 1872–1880. doi: 10.3748/wjg.v23.i10.1872
|
| [5] | K. Ohno, F. Hata, H. Nishimori, T. Yasoshima, Y. Yanai, K. Sogahata, et al., Metastatic-associated biological properties and differential gene expression profiles in established highly liver and peritoneal metastatic cell lines of human pancreatic cancer, J. Exp. Clin. Cancer Res., 22 (2003), 623–631. |
| [6] | S. Yachida, C. A. Iacobuzio-Donahue, The pathology and genetics of metastatic pancreatic cancer, Arch. Pathol. Lab. Med., 133 (2009), 413–422. |
| [7] |
A. M. Krebs, J. Mitschke, M. L. Losada, O. Schmalhofer, M. Boerries, H. Busch, et al., The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer, Nat. Cell Biol., 19 (2017), 518–529. doi: 10.1038/ncb3513
|
| [8] |
C. Moriya, H. Taniguchi, K. Miyata, N. Nishiyama, K. Kataoka, K. Imai, Inhibition of PRDM14 expression in pancreatic cancer suppresses cancer stem-like properties and liver metastasis in mice, Carcinogenesis, 38 (2017), 638–648. doi: 10.1093/carcin/bgx040
|
| [9] |
J. A. Galvan, I. Zlobec, M. Wartenberg, A. Lugli, B. Gloor, A. Perren, et al., Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer, Br. J. Cancer, 112 (2015), 1944–1950. doi: 10.1038/bjc.2015.177
|
| [10] |
H. Knaack, L. Lenk, L. M. Philipp, L. Miarka, S. Rahn, F. Viol, et al., Liver metastasis of pancreatic cancer: the hepatic microenvironment impacts differentiation and self-renewal capacity of pancreatic ductal epithelial cells, Oncotarget, 9 (2018), 31771–31786. doi: 10.18632/oncotarget.25884
|
| [11] |
V. Quaranta, C. Rainer, S. R. Nielsen, M. L. Raymant, M. S. Ahmed, D. D. Engle, et al., Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer, Cancer Res., 78 (2018), 4253–4269. doi: 10.1158/0008-5472.CAN-17-3876
|
| [12] |
H. Griesmann, C. Drexel, N. Milosevic, B. Sipos, J. Rosendahl, T. M. Gress, et al., Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer, Gut, 66 (2017), 1278–1285. doi: 10.1136/gutjnl-2015-310049
|
| [13] |
F. Runa, S. Hamalian, K. Meade, P. Shisgal, P. C. Gray, J. A. Kelber, Tumor microenvironment heterogeneity: challenges and opportunities, Curr. Mol. Biol. Rep., 3 (2017), 218–229. doi: 10.1007/s40610-017-0073-7
|
| [14] | Cancer Genome Atlas Research Network, Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma, Cancer Cell, 32 (2017), 185–203. |
| [15] |
R. A. Moffitt, R. Marayati, E. L. Flate, K. E. Volmar, S. G. Loeza, K. A. Hoadley, et al., Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma, Nat. Genet., 47 (2015), 1168–1178. doi: 10.1038/ng.3398
|
| [16] |
F. Puleo, R. Nicolle, Y. Blum, J. Cros, L. Marisa, P. Demetter, et al., Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features, Gastroenterology, 155 (2018), 1999–2013. doi: 10.1053/j.gastro.2018.08.033
|
| [17] |
J. Peng, B. F. Sun, C. Y. Chen, J. Y. Zhou, Y. S. Chen, H. Chen, et al., Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma, Cell Res., 29 (2019), 725–738. doi: 10.1038/s41422-019-0195-y
|
| [18] |
G. Yu, L. G. Wang, Y. Han, Q. Y. He, ClusterProfiler: an R package for comparing biological themes among gene clusters, OMICS, 16 (2012), 284–287. doi: 10.1089/omi.2011.0118
|
| [19] |
D. A. Barbie, P. Tamayo, J. S. Boehm, S. Y. Kim, S. E. Moody, I. F. Dunn, et al., Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1, Nature, 462 (2009), 108–112. doi: 10.1038/nature08460
|
| [20] |
A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, et al., Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci. U. S. A., 102 (2005), 15545–15550. doi: 10.1073/pnas.0506580102
|
| [21] |
S. Hanzelmann, R. Castelo, J. Guinney, GSVA: gene set variation analysis for microarray and RNA-seq data, BMC Bioinf., 14 (2013), 7. doi: 10.1186/1471-2105-14-7
|
| [22] | T. M. Therneau, T. Lumley, A. Elizabeth, C. Cynthia, Survival: Survival Analysis, 2020. Available from: https://cran.r-project.org/web/packages/survival/index.html. |
| [23] |
L. Phan, P. C. Chou, G. Velazquez-Torres, I. Samudio, K. Parreno. Y. Huang, et al., The cell cycle regulator 14-3-3sigma opposes and reverses cancer metabolic reprogramming, Nat. Commun., 6 (2015), 7530. doi: 10.1038/ncomms8530
|
| [24] | C. A. Rimland, S. G. Tilson, C. M. Morell, R. A. Tomaz, W. Y. Lu, S. E. Adams, et al., Regional differences in human biliary tissues and corresponding in vitro derived organoids, Hepatology, 2020 (2020). |
| [25] |
Y. Senbabaoglu, R. S. Gejman, A. G. Winer, M. Liu, E. M. Van Allen, G. de Velasco, et al., Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures, Genome Biol., 17 (2016), 231. doi: 10.1186/s13059-016-1092-z
|
| [26] |
F. Fu, S. Liu, S. Zeng, H. Shen, The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma, Mol. Cancer, 17 (2018), 62. doi: 10.1186/s12943-018-0815-z
|
| [27] |
T. Y. S. Le Large, M. F. Bijlsma, G. Kazemier, H. W. M. van Laarhoven, E. Giovannetti, C. R. Jimenez, Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies, Semin. Cancer Biol., 44 (2017), 153–169. doi: 10.1016/j.semcancer.2017.03.008
|
| [28] |
K. T. Yeung, J. Yang, Epithelial-mesenchymal transition in tumor metastasis, Mol. Oncol., 11 (2017), 28–39. doi: 10.1002/1878-0261.12017
|
| [29] |
E. Rodriguez-Aznar, L. Wiesmuller, B. Sainz, P. C. Hermann, EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells, Cancers (Basel), 11 (2019), 1136. doi: 10.3390/cancers11081136
|
| [30] |
J. Yang, B. Ren, G. Yang, H. Wang, G. Chen, L. You, et al., The enhancement of glycolysis regulates pancreatic cancer metastasis, Cell. Mol. Life Sci., 77 (2020), 305–321. doi: 10.1007/s00018-019-03278-z
|
| [31] |
F. Robin, G. Angenard, L. Cano, L. Courtin-Tanguy, E. Gaignard, Z. E. Khene, et al., Molecular profiling of stroma highlights stratifin as a novel biomarker of poor prognosis in pancreatic ductal adenocarcinoma, Br. J. Cancer, 123 (2020), 72–80. doi: 10.1038/s41416-020-0863-1
|
| [32] | H. Yao, Z. Yang, Z. Liu, X. Miao, L. Yang, D. Li, et al., Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma, Cancer Biomark, 17 (2016), 397–404. |
| [33] |
M. Fornaro, R. Dell'Arciprete, M. Stella, C. Bucci, M. Nutini, M. G. Capri, et al., Cloning of the gene encoding Trop-2, a cell-surface glycoprotein expressed by human carcinomas, Int. J. Cancer, 62 (1995), 610–618. doi: 10.1002/ijc.2910620520
|
| [34] |
M. Tanaka, A. L. Mihaljevic, P. Probst, M. Heckler, U. Klaiber, U. Heger, et al., Meta-analysis of recurrence pattern after resection for pancreatic cancer, Br. J. Surg., 106 (2019), 1590–1601. doi: 10.1002/bjs.11295
|
| [35] |
M. Najafi, N. Hashemi Goradel, B. Farhood, E. Salehi, M. S. Nashtaei, N. Khanlarkhani, et al., Macrophage polarity in cancer: A review, J. Cell. Biochem., 120 (2019), 2756–2765. doi: 10.1002/jcb.27646
|
| [36] | W. Li, J. Zeng, B. Luo, Y. Mao, Y. Liang, W. Zhao, et al., High expression of activated CD4(+) memory T cells and CD8(+) T cells and low expression of M0 macrophage are associated with better clinical prognosis in bladder cancer patients, Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi, 36 (2020), 97–103. |
 mbe-18-02-088-s001.xlsx mbe-18-02-088-s001.xlsx |
 |
| mbe-18-02-088-s002.xlsx |
|
Figures(5) / Tables(2)

Yang Yu, Zhe Wang, Dai hai Mo, Zhen Wang, Gang Li. Transcriptome profiling reveals liver metastasis-associated genes in pancreatic ductal adenocarcinoma[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1708-1721. doi: 10.3934/mbe.2021088







 DownLoad:
DownLoad: