Citation: Xiaochen Fan, David A F Loebel, Heidi Bildsoe, Emilie E Wilkie, Jing Qin, Junwen Wang, Patrick P L Tam. Tissue interactions, cell signaling and transcriptional control in the cranial mesoderm during craniofacial development[J]. AIMS Genetics, 2016, 3(1): 74-98. doi: 10.3934/genet.2016.1.74

| [1] | Brault V, Moore R, Kutsch S, et al. (2001) Inactivation of the beta-catenin gene by {Wnt1-Cre-mediated} deletion results in dramatic brain malformation and failure of craniofacial development. Development 128: 1253-1264. |
| [2] |
Noden DM, Trainor PA (2005) Relations and interactions between cranial mesoderm and neural crest populations. J Anat 207: 575-601. doi: 10.1111/j.1469-7580.2005.00473.x

|
| [3] |
Trainor P, Krumlauf R (2000) Plasticity in mouse neural crest cells reveals a new patterning role for cranial mesoderm. Nat cell biol 2: 96-9102. doi: 10.1038/35000051
|
| [4] | Trainor PA, Tam PP (1995) Cranial paraxial mesoderm and neural crest cells of the mouse embryo: co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development 121: 2569-2582. |
| [5] |
McBratney-Owen, Iseki S, Bamforth SD (2008) Development and tissue origins of the mammalian cranial base. Dev Biol 322: 121-132. doi: 10.1016/j.ydbio.2008.07.016
|
| [6] |
Lescroart F, Kelly RG, Le Garrec JF, et al. (2010) Clonal analysis reveals common lineage relationships between head muscles and second heart field derivatives in the mouse embryo. Development 137: 3269-3279. doi: 10.1242/dev.050674
|
| [7] | Yoshida T, Vivatbutsiri P, Gillian MK, et al. (2008) Cell lineage in mammalian craniofacial mesenchyme. Mech dev 125: 797-808. |
| [8] | Lee Y-HH, Saint-Jeannet J-PP (2011) Sox9 function in craniofacial development and disease. Genesis 49: 200-208. |
| [9] | Bildsoe H, Loebel DAF, Jones VJ, et al. (2009) Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev Biol 331: 176-188. |
| [10] | Bothe I, Tenin G, Oseni A, et al. (2011) Dynamic control of head mesoderm patterning. Development 138: 2807-2821. |
| [11] | Tam PPL, Meier S (1982) The establishment of a somitomeric pattern in the mesoderm of the gastrulating mouse embryo. Am J Anat 164: 209-225. |
| [12] |
Meier S, Tam PPL (1982) Metameric pattern development in the embryonic axis of the mouse I. Differentiation 21: 95-108. doi: 10.1111/j.1432-0436.1982.tb01202.x
|
| [13] |
Tzahor E, Kempf H, Mootoosamy RC, et al. (2003) Antagonists of Wnt and {BMP} signaling promote the formation of vertebrate head muscle. Genes dev 17: 3087-3099. doi: 10.1101/gad.1154103
|
| [14] | Michailovici I, Eigler T, Tzahor E (2015) Craniofacial Muscle Development. Curr top dev biol 115: 3-30. |
| [15] |
Tzahor E (2015) Head muscle development. Results and problems in cell differentiation 56: 123-142. doi: 10.1007/978-3-662-44608-9_6
|
| [16] |
Tajbakhsh S, Cossu G (1997) Establishing myogenic identity during somitogenesis. Curr Opin Genet Dev 7: 634-641. doi: 10.1016/S0959-437X(97)80011-1
|
| [17] |
Harel I, Nathan E, Tirosh-Finkel L, et al. (2009) Distinct origins and genetic programs of head muscle satellite cells. Dev cell 16: 822-832. doi: 10.1016/j.devcel.2009.05.007
|
| [18] | Kitamura K, Miura H, S M-T, et al. (1999) Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 126: 5749-5758. |
| [19] |
Dong F, Sun X, Liu W, et al. (2006) Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development 133: 4891-4899. doi: 10.1242/dev.02693
|
| [20] |
Diehl AG, Zareparsi S, Qian M, et al. (2006) Extraocular muscle morphogenesis and gene expression are regulated by Pitx2 gene dose. Invest Ophthalmol Vis Sci 47: 1785-1793. doi: 10.1167/iovs.05-1424
|
| [21] |
Richardson L, Venkataraman S, Stevenson P, et al. (2014) EMAGE mouse embryo spatial gene expression database: 2014 update. Nucleic acids res 42: D835-844. doi: 10.1093/nar/gkt1155
|
| [22] |
Zhang Z, Huynh T, Baldini A (2006) Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 133: 3587-3595. doi: 10.1242/dev.02539
|
| [23] |
Aggarwal VS, Carpenter C, Freyer L, et al. (2010) Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev Biol 344: 669-681. doi: 10.1016/j.ydbio.2010.05.496
|
| [24] | Franco HL, Casasnovas J, Rodríguez-Medina JR, et al. (2011) Redundant or separate entities? - Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 39: 1177-1186. |
| [25] | Hebrok M, Wertz K, Fuchtbauer EM (1994) M-twist is an inhibitor of muscle differentiation. Dev biol 165: 537-544. |
| [26] | Spicer DB, Rhee J, Cheung WL, et al. (1996) Inhibition of myogenic {bHLH} and {MEF2} transcription factors by the {bHLH} protein Twist. Science 272: 1476-1480. |
| [27] | Bildsoe H, Loebel DA, Jones VJ, et al. (2013) The mesenchymal architecture of the cranial mesoderm of mouse embryos is disrupted by the loss of Twist1 function. Dev Biol 374: 295-307. |
| [28] |
Goodnough LH, Dinuoscio GJ, Atit RP (2016) Twist1 contributes to cranial bone initiation and dermal condensation by maintaining wnt signaling responsiveness. Dev dyns 245: 144-156. doi: 10.1002/dvdy.24367
|
| [29] | Longabaugh WJR, Davidson EH, Bolouri H (2009) Visualization, documentation, analysis, and communication of large-scale gene regulatory networks. Biochim Biophysica Acta 1789: 363-374. |
| [30] |
Katsuno Y, Lamouille S, Derynck R (2013) TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr opin oncol 25: 76-84. doi: 10.1097/CCO.0b013e32835b6371
|
| [31] |
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat rev Mol cell biol 15: 178-196. doi: 10.1038/nrm3758
|
| [32] |
Diogo R, Kelly RG, Christiaen L, et al. (2015) A new heart for a new head in vertebrate cardiopharyngeal evolution. Nature 520: 466-473. doi: 10.1038/nature14435
|
| [33] |
Lewis AE, Vasudevan HN, O’Neill AK, et al. (2013) The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev biol 379: 229-234. doi: 10.1016/j.ydbio.2013.04.026
|
| [34] | Wang X-HH, Wang Y, Liu AKK, et al. (2015) Genome-wide identification and analysis of basic helix-loop-helix domains in dog, Canis lupus familiaris. Mol genet genomics 290: 633-648. |
| [35] |
Liem KF, Tremml G, Roelink H, et al. (1995) Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell 82: 969-979. doi: 10.1016/0092-8674(95)90276-7
|
| [36] | Kanzler B, Foreman RK, Labosky PA, et al. (2000) BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development 127: 1095-1104. |
| [37] |
Dudas M, Sridurongrit S, Nagy A, et al. (2004) Craniofacial defects in mice lacking {BMP} type I receptor Alk2 in neural crest cells. Mech dev 121: 173-182. doi: 10.1016/j.mod.2003.12.003
|
| [38] |
Dudas M, Kim J, Li W-YY, et al. (2006) Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev biol 296: 298-314. doi: 10.1016/j.ydbio.2006.05.030
|
| [39] |
Liu W, Selever J, Murali D, et al. (2005) Threshold-specific requirements for Bmp4 in mandibular development. Dev biol 283: 282-293. doi: 10.1016/j.ydbio.2005.04.019
|
| [40] |
Ko SO, Chung IH, Xu X, et al. (2007) Smad4 is required to regulate the fate of cranial neural crest cells. Dev Biol 312: 435-447. doi: 10.1016/j.ydbio.2007.09.050
|
| [41] |
Zhao H, Oka K, Bringas P, et al. (2008) {TGF-β} type I receptor Alk5 regulates tooth initiation and mandible patterning in a type {II} receptor-independent manner. Dev Biol 320: 19-29. doi: 10.1016/j.ydbio.2008.03.045
|
| [42] | Roybal PG, Wu NL, Sun J, et al. (2010) Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev biol 343: 28-39. |
| [43] | Kim H, Rice D, Kettunen P, et al. (1998) FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125: 1241-1251. |
| [44] | Bonilla-Claudio M, Wang J, Bai Y, et al. (2012) Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development 139: 709-719. |
| [45] |
Lu MF, Pressman C, Dyer R, et al. (1999) Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 401: 276-278. doi: 10.1038/45797
|
| [46] | Tapscott SJ, Weintraub H (1991) {MyoD} and the regulation of myogenesis by helix-loop-helix proteins. J clin invest 87: 1133-1138. |
| [47] | Langlands K, Yin X, Anand G, et al. (1997) Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J biol chem 272: 19785-19793. |
| [48] |
Nie X, Luukko K, Kettunen P (2006) {FGF} signalling in craniofacial development and developmental disorders. Oral Dis 12: 102-111. doi: 10.1111/j.1601-0825.2005.01176.x
|
| [49] | Suzuki A, Sangani DR, Ansari A (2015) Molecular mechanisms of midfacial developmental defects. Dev Dyn 245: 276-293. |
| [50] | Bachler M, Neubüser A (2001) Expression of members of the Fgf family and their receptors during midfacial development. Mech dev 100: 313-316. |
| [51] |
Wilkie AOM (1997) Craniosynostosis: genes and mechanisms. Hum Mol Genet 6: 1647-1656. doi: 10.1093/hmg/6.10.1647
|
| [52] | Abu-Issa R, Smyth G, Smoak I, et al. (2002) Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 129: 4613-4625. |
| [53] |
Griffin JN, Compagnucci C, Hu D, et al. (2013) Fgf8 dosage determines midfacial integration and polarity within the nasal and optic capsules. Dev biol 374: 185-197. doi: 10.1016/j.ydbio.2012.11.014
|
| [54] |
Wang C, Chang JY, Yang C, et al. (2013) Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J biol chem 288: 22174-22183. doi: 10.1074/jbc.M113.463620
|
| [55] |
Kasberg AD, Brunskill EW, Potter SS (2013) {SP8} regulates signaling centers during craniofacial development. Dev Biol 381: 312-323. doi: 10.1016/j.ydbio.2013.07.007
|
| [56] | Yang X, Kilgallen S, Andreeva V, et al. (2010) Conditional expression of Spry1 in neural crest causes craniofacial and cardiac defects. Dev biol 10: 48. |
| [57] | Yu K, Xu J, Liu Z, et al. (2003) Conditional inactivation of {FGF} receptor 2 reveals an essential role for {FGF} signaling in the regulation of osteoblast function and bone growth. Development 130: 3063-3074. |
| [58] |
Holmes G, Basilico C (2012) Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev Biol 368: 283-293. doi: 10.1016/j.ydbio.2012.05.026
|
| [59] |
Stottmann RW, Anderson RM, Klingensmith J (2001) The BMP antagonists Chordin and Noggin have essential but redundant roles in mouse mandibular outgrowth. Dev Biol 240: 457-473. doi: 10.1006/dbio.2001.0479
|
| [60] | Tucker A, Sharpe P (2004) The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet 5: 499-508. |
| [61] | Tirosh-Finkel L, Zeisel A, Brodt-Ivenshitz M, et al. (2010) BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 137: 2989-3000. |
| [62] | Olwin BB, Rapraeger A (1992) Repression of myogenic differentiation by aFGF, bFGF, and K-FGF is dependent on cellular heparan sulfate. J cell biol 118: 631-639. |
| [63] |
von Scheven G, Alvares LEE, Mootoosamy RC, et al. (2006) Neural tube derived signals and Fgf8 act antagonistically to specify eye versus mandibular arch muscles. Development 133: 2731-2745. doi: 10.1242/dev.02426
|
| [64] | Michailovici I, Harrington HA, Azogui HH, et al. (2014) Nuclear to cytoplasmic shuttling of ERK promotes differentiation of muscle stem/progenitor cells. Development 141: 2611-2620. |
| [65] | Frank DU, Fotheringham LK, Brewer JA, et al. (2002) An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 129: 4591-4603. |
| [66] | Rinon A, Lazar S, Marshall H, et al. (2007) Cranial neural crest cells regulate head muscle patterning and differentiation during vertebrate embryogenesis. Development 134: 3065-3075. |
| [67] | Mani P, Jarrell A, Myers J, et al. (2010) Visualizing canonical Wnt signaling during mouse craniofacial development. Dev Dyn 239: 354-363. |
| [68] | Hari L, Brault V, Kléber M, et al. (2002) Lineage-specific requirements of beta-catenin in neural crest development. J cell biol 159: 867-880. |
| [69] |
Goodnough LH, Chang AT, Treloar C, et al. (2012) Twist1 mediates repression of chondrogenesis by β-catenin to promote cranial bone progenitor specification. Development 139: 4428-4438. doi: 10.1242/dev.081679
|
| [70] | Goodnough LH, Dinuoscio GJ, Ferguson JW, et al. (2014) Distinct requirements for cranial ectoderm and mesenchyme-derived wnts in specification and differentiation of osteoblast and dermal progenitors. Genetics 10: e1004152. |
| [71] |
Wang Y, Song L, Zhou CJ (2011) The canonical Wnt/β-catenin signaling pathway regulates Fgf signaling for early facial development. Dev biol 349: 250-260. doi: 10.1016/j.ydbio.2010.11.004
|
| [72] |
Reid BS, Yang H, Melvin VS, et al. (2011) Ectodermal Wnt/β-catenin signaling shapes the mouse face. Dev biol 349: 261-269. doi: 10.1016/j.ydbio.2010.11.012
|
| [73] |
Klaus A, Saga Y, Taketo MM, et al. (2007) Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci U S A 104: 18531-18536. doi: 10.1073/pnas.0703113104
|
| [74] | Wang XP, O'Connell DJ, Lund JJ, et al. (2009) Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development 136: 1939-1949. |
| [75] |
Day TF, Guo X, Lisa G-B, et al. (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev cell 8: 739-750. doi: 10.1016/j.devcel.2005.03.016
|
| [76] | Reinhold MI, Kapadia RM, Liao Z, et al. (2006) The Wnt-inducible transcription factor Twist1 inhibits chondrogenesis. J biol chem 281: 1381-1388. |
| [77] |
Hoang BH, Thomas JT, Abdul-Karim FW, et al. (1998) Expression pattern of two Frizzled-related genes, Frzb-1 and Sfrp-1, during mouse embryogenesis suggests a role for modulating action of Wnt family members. Dev Dyn 212: 364-372. doi: 10.1002/(SICI)1097-0177(199807)212:3<364::AID-AJA4>3.0.CO;2-F
|
| [78] |
Lescher B, Haenig B, Kispert A (1998) sFRP-2 is a target of the Wnt-4 signaling pathway in the developing metanephric kidney. Dev Dyn 213: 440-451. doi: 10.1002/(SICI)1097-0177(199812)213:4<440::AID-AJA9>3.0.CO;2-6
|
| [79] |
Yamashita S, Andoh M, Ueno-Kudoh H, et al. (2009) Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp Cell Res 315: 2231-2240. doi: 10.1016/j.yexcr.2009.03.008
|
| [80] |
Eames BF, Sharpe PT, Helms JA (2004) Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Dev Biol 274: 188-200. doi: 10.1016/j.ydbio.2004.07.006
|
| [81] |
Zhou G, Zheng Q, Engin F, et al. (2006) Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A 103: 19004-19009. doi: 10.1073/pnas.0605170103
|
| [82] | Akiyama H, Lyons JP, Yuko M-A, et al. (2004) Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes dev 18: 1072-1087. |
| [83] | Ting M-CC, Wu NL, Roybal PG, et al. (2009) EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 136: 855-864. |
| [84] | Chang AT, Liu Y, Ayyanathan K, et al. (2015) An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes dev 29: 603-616. |
| [85] | Lee H-YY, Kléber M, Hari L, et al. (2004) Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science 303: 1020-1023. |
| [86] | Chen ZF, Behringer RR (1995) Twist Is Required in Head Mesenchyme for Cranial Neural Tube Morphogenesis. Genes Dev 9: 686-699. |
| [87] |
Soo K, P OR, Meredith, Khoo P-LL, et al. (2002) Twist function is required for the morphogenesis of the cephalic neural tube and the differentiation of the cranial neural crest cells in the mouse embryo. Dev biol 247: 251-270. doi: 10.1006/dbio.2002.0699
|
| [88] |
Gitelman I (1997) Twist protein in mouse embryogenesis. Dev biol 189: 205-214. doi: 10.1006/dbio.1997.8614
|
| [89] |
Meng T, Huang Y, Wang S, et al. (2015) Twist1 Is Essential for Tooth Morphogenesis and Odontoblast Differentiation. J Biol Chem 290: 29593-29602. doi: 10.1074/jbc.M115.680546
|
| [90] |
Loebel DA, Hor AC, Bildsoe HK, et al. (2014) Timed deletion of Twist1 in the limb bud reveals age-specific impacts on autopod and zeugopod patterning. PLoS One 9: e98945. doi: 10.1371/journal.pone.0098945
|
| [91] |
Zhang Y, Blackwell EL, T M, Mitchell, et al. (2012) Specific inactivation of Twist1 in the mandibular arch neural crest cells affects the development of the ramus and reveals interactions with hand2. Dev dyn 241: 924-940. doi: 10.1002/dvdy.23776
|
| [92] | Morris-Wiman J, Brinkley LL (1990) The role of the mesenchyme in mouse neural fold elevation. I. Patterns of mesenchymal cell distribution and proliferation in embryos developing in vitro. Am J anat 188: 121-132. |
| [93] |
Teng Y, Li X (2014) The roles of HLH transcription factors in epithelial mesenchymal transition and multiple molecular mechanisms. Clin Exp Metastasis 31: 367-377. doi: 10.1007/s10585-013-9621-6
|
| [94] |
Lee K-WW, Lee NK, Ham S, et al. (2014) Twist1 is essential in maintaining mesenchymal state and tumor-initiating properties in synovial sarcoma. Cancer lett 343: 62-73. doi: 10.1016/j.canlet.2013.09.013
|
| [95] | Tsai JH, Donaher JL, Murphy DA, et al. (2012) Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell 22: 725-736. |
| [96] | Szklarczyk D, Franceschini A, Wyder S (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic acids Res 43: D447-452. |
| [97] | Csardi G, Nepusz T (2006) The igraph software package for complex network research. Inter Journal. |
| [98] |
Takada R, Satomi Y, Kurata T, et al. (2006) Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev cell 11: 791-801. doi: 10.1016/j.devcel.2006.10.003
|
| [99] |
Grenier J, Teillet M-AA, Grifone R, et al. (2009) Relationship between neural crest cells and cranial mesoderm during head muscle development. PloS one 4: e4381. doi: 10.1371/journal.pone.0004381
|
| [100] | Schneider RA (1999) Neural crest can form cartilages normally derived from mesoderm during development of the avian head skeleton. Dev biol 208: 441-455. |
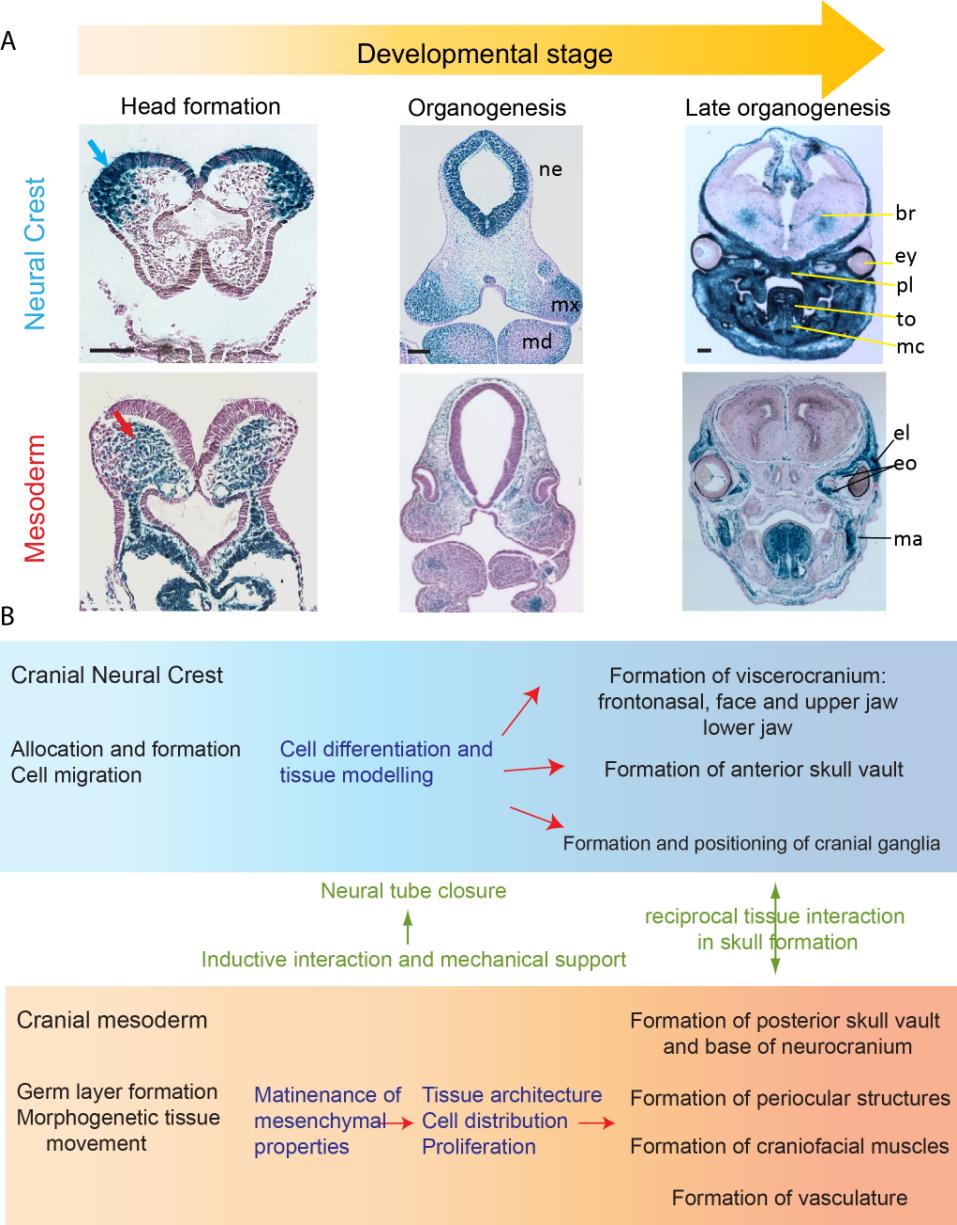
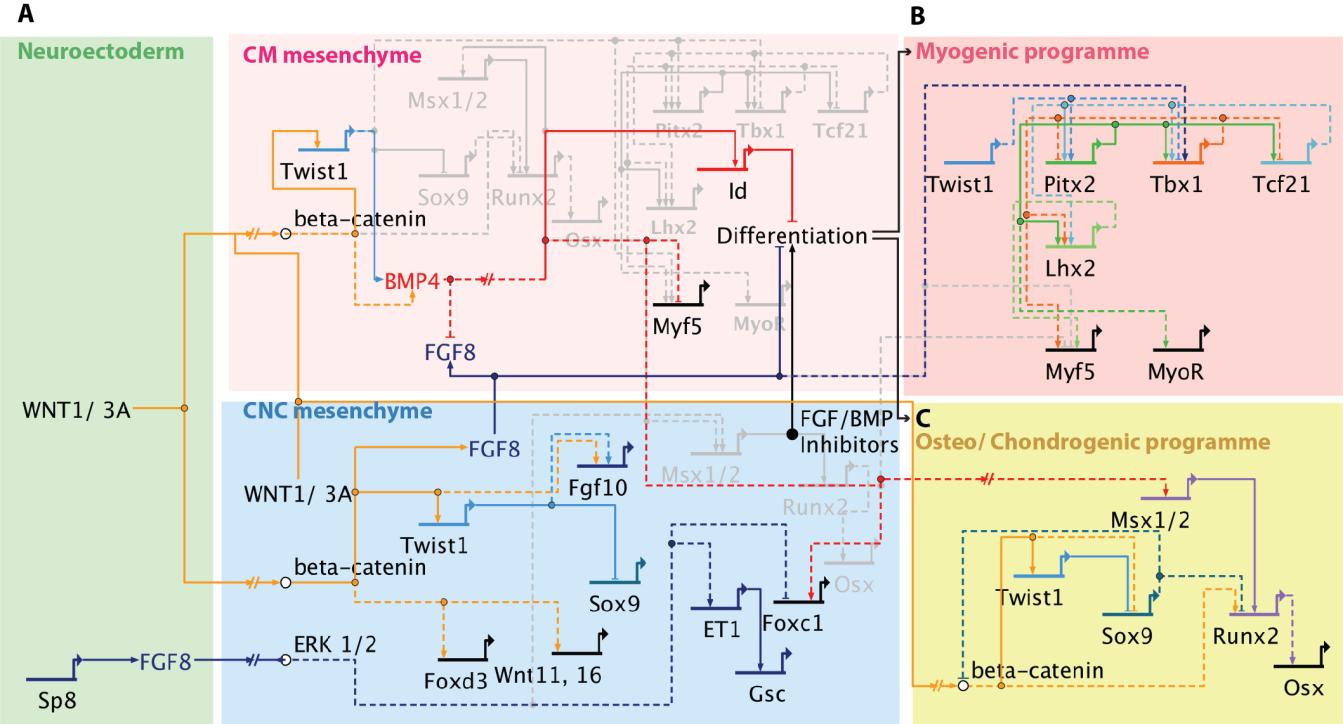
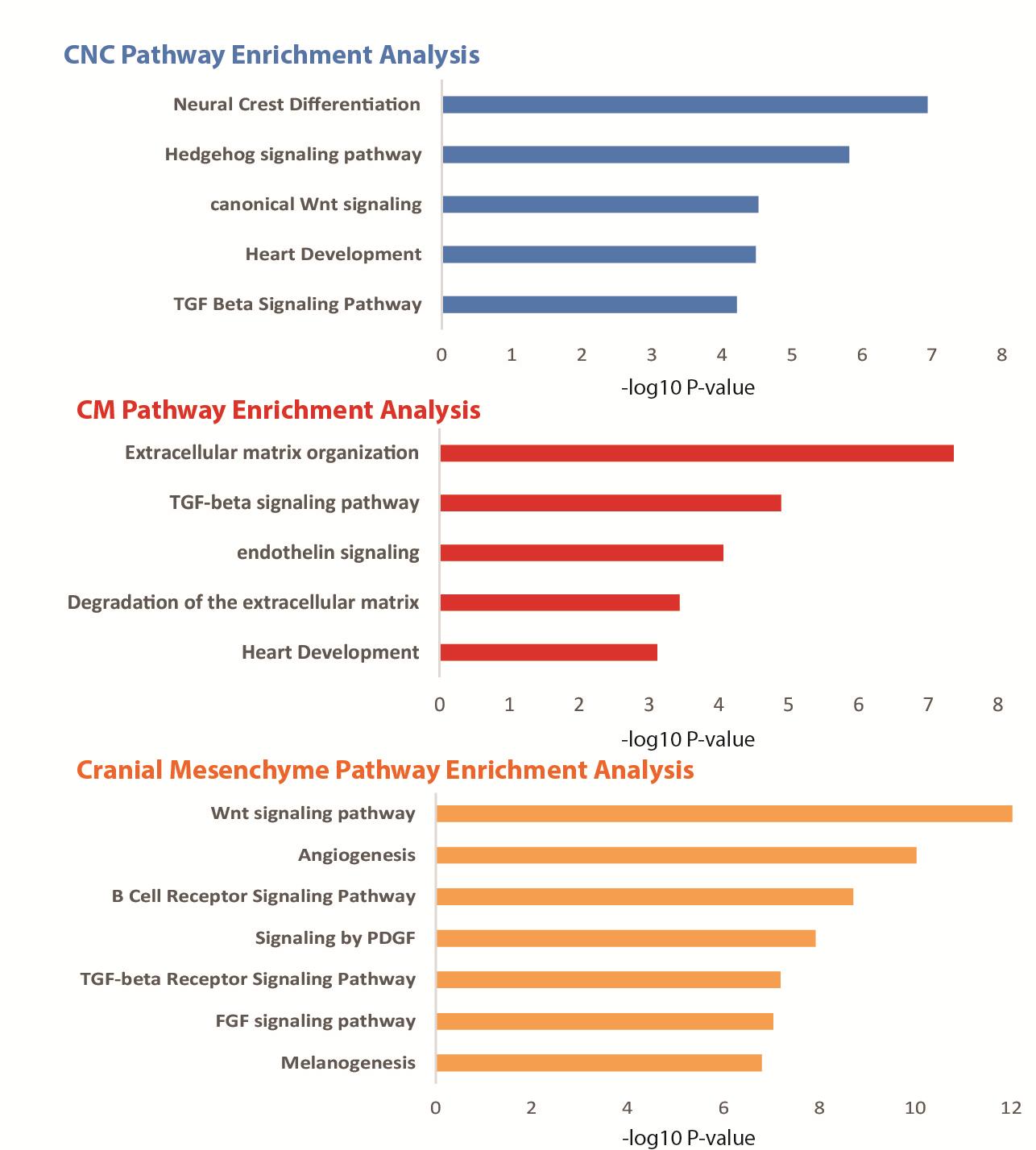
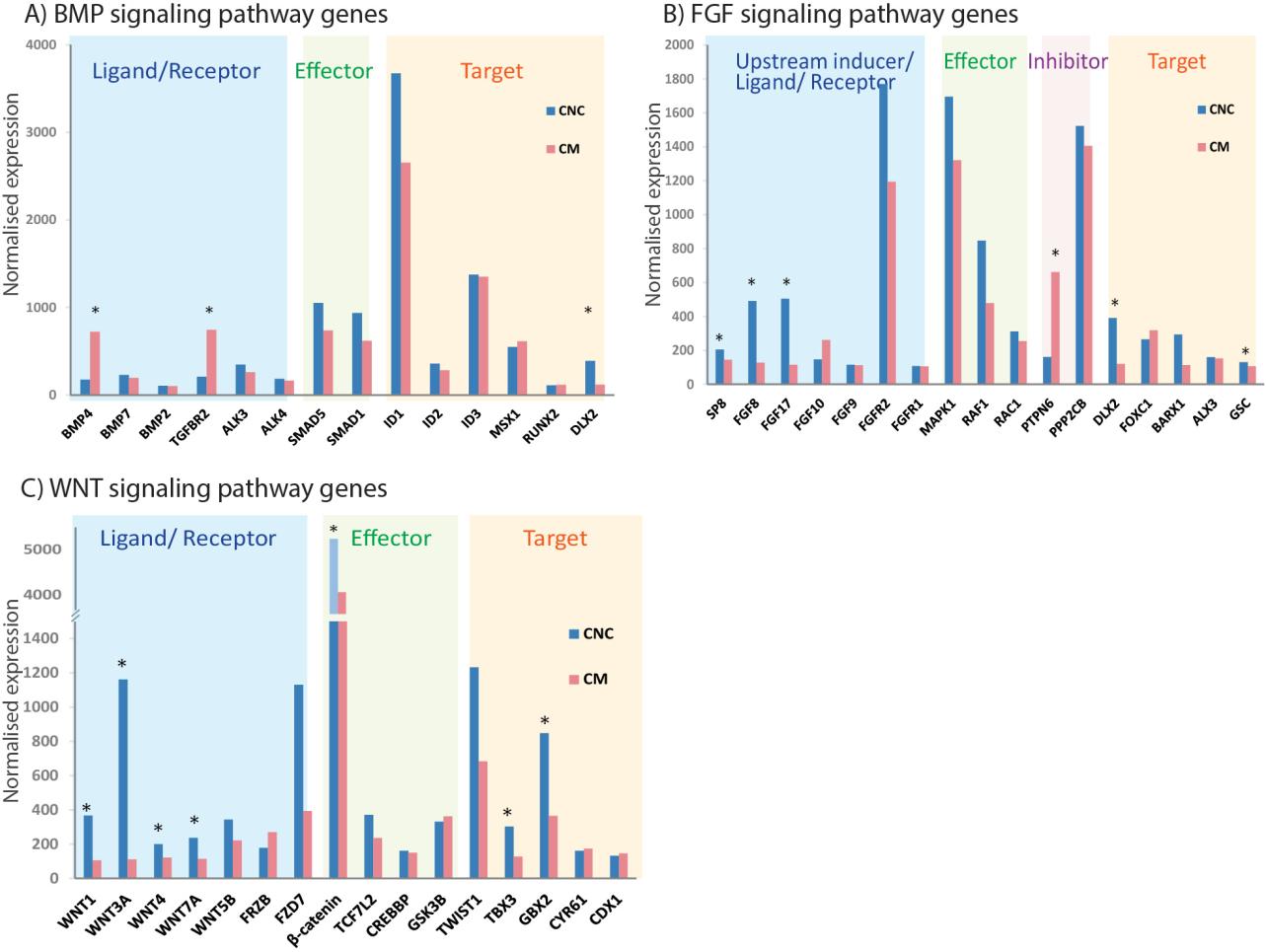
Figures(6) / Tables(1)

Xiaochen Fan, David A F Loebel, Heidi Bildsoe, Emilie E Wilkie, Jing Qin, Junwen Wang, Patrick P L Tam. Tissue interactions, cell signaling and transcriptional control in the cranial mesoderm during craniofacial development[J]. AIMS Genetics, 2016, 3(1): 74-98. doi: 10.3934/genet.2016.1.74







 DownLoad:
DownLoad: