Citation: Jue Er Amanda Lee, Linda May Parsons, Leonie M. Quinn. MYC function and regulation in flies: how Drosophila has enlightened MYC cancer biology[J]. AIMS Genetics, 2014, 1(1): 81-98. doi: 10.3934/genet.2014.1.81

| [1] | Vennstrom B, Sheiness D, Zabielski J, et al. (1982) Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol 42: 773-779. |
| [2] |
Bouchard C, Staller P, Eilers M (1998) Control of cell proliferation by Myc. Trends Cell Biol 8: 202-206. doi: 10.1016/S0962-8924(98)01251-3

|
| [3] |
Schmidt EV (1999) The role of c-myc in cellular growth control. Oncogene 18: 2988-2996. doi: 10.1038/sj.onc.1202751
|
| [4] |
Trumpp A, Refaeli Y, Oskarsson T, et al. (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768-773. doi: 10.1038/414768a
|
| [5] |
Dang CV (2010) Enigmatic MYC Conducts an Unfolding Systems Biology Symphony. Genes Cancer 1: 526-531. doi: 10.1177/1947601910378742
|
| [6] |
Gallant P, Shiio Y, Cheng PF, et al. (1996) Myc and Max homologs in Drosophila. Science 274: 1523-1527. doi: 10.1126/science.274.5292.1523
|
| [7] |
Johnston LA, Prober DA, Edgar BA, et al. (1999) Drosophila myc regulates cellular growth during development. Cell 98: 779-790. doi: 10.1016/S0092-8674(00)81512-3
|
| [8] |
Pierce SB, Yost C, Britton JS, et al. (2004) dMyc is required for larval growth and endoreplication in Drosophila. Development 131: 2317-2327. doi: 10.1242/dev.01108
|
| [9] |
Grewal SS, Li L, Orian A, et al. (2005) Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol 7: 295-302. doi: 10.1038/ncb1223
|
| [10] |
Schreiber-Agus N, Stein D, Chen K, et al. (1997) Drosophila Myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc Natl Acad Sci U S A 94: 1235-1240. doi: 10.1073/pnas.94.4.1235
|
| [11] |
Benassayag C, Montero L, Colombie N, et al. (2005) Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol Cell Biol 25: 9897-9909. doi: 10.1128/MCB.25.22.9897-9909.2005
|
| [12] |
Laurenti E, Varnum-Finney B, Wilson A, et al. (2008) Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 3: 611-624. doi: 10.1016/j.stem.2008.09.005
|
| [13] |
Blackwood EM, Eisenman RN (1991) Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251: 1211-1217. doi: 10.1126/science.2006410
|
| [14] |
Amati B, Dalton S, Brooks MW, et al. (1992) Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359: 423-426. doi: 10.1038/359423a0
|
| [15] |
Amati B, Land H (1994) Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Genet Dev 4: 102-108. doi: 10.1016/0959-437X(94)90098-1
|
| [16] | Grandori C, Mac J, Siebelt F, et al. (1996) Myc-Max heterodimers activate a DEAD box gene and interact with multiple E box-related sites in vivo. EMBO J, 15: 4344-4357. |
| [17] |
Eilers M, Eisenman RN (2008) Myc's broad reach. Genes Dev 22: 2755-2766. doi: 10.1101/gad.1712408
|
| [18] |
Steiger D, Furrer M, Schwinkendorf D (2008) Max-independent functions of Myc in Drosophila melanogaster. Nat Genet 40: 1084-1091. doi: 10.1038/ng.178
|
| [19] | Bellosta P, Hulf T, Balla Diop S, et al. (2008) Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc Natl Acad Sci U S A 102: 11799-11804. |
| [20] |
Wood MA, McMahon SB, Cole MD (2000) An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell 5: 321-330. doi: 10.1016/S1097-2765(00)80427-X
|
| [21] |
Rahl PB, Lin CY, Seila AC, et al. (2010) c-Myc Regulates Transcriptional Pause Release. Cell 141: 432-445. doi: 10.1016/j.cell.2010.03.030
|
| [22] |
Wasylishen AR, Penn LZ (2010) Myc: The Beauty and the Beast. Genes Cancer 1: 532-541. doi: 10.1177/1947601910378024
|
| [23] |
Orian A, Grewal SS, Knoepfler PS, et al. (2005) Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harb Sym 70: 299-307. doi: 10.1101/sqb.2005.70.019
|
| [24] |
Orian A, van Steensel B, Delrow J, et al. (2003) Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev 17: 1101-1114. doi: 10.1101/gad.1066903
|
| [25] |
Grandori C, Cowley SM, James LP, et al. (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Bi 16: 653-699. doi: 10.1146/annurev.cellbio.16.1.653
|
| [26] |
Coller HA, Grandori C, Tamayo P, et al. (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A 97: 3260-3265. doi: 10.1073/pnas.97.7.3260
|
| [27] |
Nie Z, Hu G, Wei G, et al. (2012) c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 151: 68-79. doi: 10.1016/j.cell.2012.08.033
|
| [28] |
Lin CY, Lovén J, Rahl PB, et al. (2012) Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 151: 56-67. doi: 10.1016/j.cell.2012.08.026
|
| [29] | Fernandez PC, Frank SR, Wang L, et al. (2003) Genomic targets of the human c-Myc protein, Genes Dev 17: 1115-1129. |
| [30] |
Levens DL (2003) Reconstructing MYC. Genes Dev 17: 1071-1077. doi: 10.1101/gad.1095203
|
| [31] |
Dang CV, O'Donnell KA, Zeller KI, et al. (2006) The c-Myc target gene network. Semin Cancer Biol 16: 253-264. doi: 10.1016/j.semcancer.2006.07.014
|
| [32] |
Ji H, Wu G, Zhan X, et al. (2011) Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 6: e26057. doi: 10.1371/journal.pone.0026057
|
| [33] | Sabò A, Amati B (2014) Genome recognition by MYC. Cold Spring Harb Perspect Med 4. |
| [34] |
Littlewood TD, Kreuzaler P, Evan GI (2012) All things to all people. Cell 151: 11-13. doi: 10.1016/j.cell.2012.09.006
|
| [35] |
Zeller KI, Zhao X, Lee CWH, et al. (2006) Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A 103: 17834-17839. doi: 10.1073/pnas.0604129103
|
| [36] |
Guccione E, Martinato F, Finocchiaro G, et al. (2006) Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol 8: 764-770. doi: 10.1038/ncb1434
|
| [37] |
Poortinga G, Wall M, Sanij E, et al. (2011) c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res 39: 3267-3281. doi: 10.1093/nar/gkq1205
|
| [38] | Poortinga G, Quinn LM, Hannan RD (2014) Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene Epub ahead of print. |
| [39] |
Barna M, Pusic A, Zollo O, et al. (2008) Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456: 971-975. doi: 10.1038/nature07449
|
| [40] |
Ruggero D (2009) The role of Myc-induced protein synthesis in cancer. Cancer Res 69: 8839-8843. doi: 10.1158/0008-5472.CAN-09-1970
|
| [41] | Ruggero D (2012) Revisiting the nucleolus: from marker to dynamic integrator of cancer signaling. Sci Signal 5: pe38. |
| [42] |
Bywater MJ, Poortinga G, Sanij E, et al. (2012) Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 22: 51-65. doi: 10.1016/j.ccr.2012.05.019
|
| [43] |
Dang CV (2012) MYC on the path to cancer. Cell 149: 22-35. doi: 10.1016/j.cell.2012.03.003
|
| [44] |
Schwab M (2004) MYCN in neuronal tumours. Cancer Lett 204: 179-187. doi: 10.1016/S0304-3835(03)00454-3
|
| [45] | Wahlström T, Arsenian Henriksson M (2014) Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta S1874-9399: 00192-00198. |
| [46] |
Dalla-Favera R, Bregni M, Erikson J, et al. (1982) Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A 79: 7824-7827. doi: 10.1073/pnas.79.24.7824
|
| [47] | Taub R, Kirsch I, Morton C, et al. (1982) Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A 79l: 7837-7841. |
| [48] |
Arabi A, Wu S, Ridderstråle K, et al. (2005) c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol 7: 303-310. doi: 10.1038/ncb1225
|
| [49] |
Grandori C, Gomez-Roman N, Felton-Edkins ZA, et al. (2005) c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 7: 311-318. doi: 10.1038/ncb1224
|
| [50] |
Schlosser I, Hölzel M, Mürnseer M, et al. (2003) A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res 31: 6148-6156. doi: 10.1093/nar/gkg794
|
| [51] |
Poortinga G, Hannan KM, Snelling H, et al. (2004) MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J 23: 3325-3335. doi: 10.1038/sj.emboj.7600335
|
| [52] |
van Riggelen J, Yetil A, Felsher DW (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev 10: 301-309. doi: 10.1038/nrc2819
|
| [53] |
Oskarsson T, Trumpp A (2005) The Myc trilogy: lord of RNA polymerases. Nat cell Biol 7: 215-217. doi: 10.1038/ncb0305-215
|
| [54] |
Gomez-Roman N, Grandori C, Eisenman RN, et al. (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290-294. doi: 10.1038/nature01327
|
| [55] |
Sanij E, Poortinga G, Sharkey K, et al. (2008) UBF levels determine the number of active ribosomal RNA genes in mammals. J Cell Biol 183: 1259-1274. doi: 10.1083/jcb.200805146
|
| [56] |
Hein N, Hannan KM, George AJ, et al. (2013) The nucleolus: an emerging target for cancer therapy. Trends Mol Med 19: 643-654. doi: 10.1016/j.molmed.2013.07.005
|
| [57] |
Giacinti C, Giordano A (2006) RB and cell cycle progression. Oncogene 25: 5220-5227. doi: 10.1038/sj.onc.1209615
|
| [58] |
Duman-Scheel M, Johnston LA, Du W (2004) Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc Natl Acad Sci U S A 101: 3857-3862. doi: 10.1073/pnas.0400526101
|
| [59] |
Maines JZ (2004) Drosophila dMyc is required for ovary cell growth and endoreplication. Development 131: 775-786. doi: 10.1242/dev.00932
|
| [60] | Wierstra I, Alves J (2008) Advances in Cancer Research, 99. Elsevier. 113-333. |
| [61] |
Levens D (2010) You Don't Muck with MYC. Genes Cancer 1: 547-554. doi: 10.1177/1947601910377492
|
| [62] |
Sears R, Leone G, DeGregori J, et al. (1999) Ras enhances Myc protein stability. Mol Cell 3: 169-179. doi: 10.1016/S1097-2765(00)80308-1
|
| [63] |
Salghetti SE, Kim SY, Tansey WP (1999) Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J 18: 717-726. doi: 10.1093/emboj/18.3.717
|
| [64] | Sears RC (2004) The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3: 1133-1137. |
| [65] |
Daneshvar K, Nath S, Khan A, et al. (2013) MicroRNA miR-308 regulates dMyc through a negative feedback loop in Drosophila. Biol Open 2: 1-9. doi: 10.1242/bio.20122725
|
| [66] |
Amati B, Sanchez-Arevalo Lobo VJ (2007) MYC degradation: deubiquitinating enzymes enter the dance. Nat Cell Biol 9: 729-731. doi: 10.1038/ncb0707-729
|
| [67] |
Yada M, Hatakeyama S, Kamura T, et al. (2004) Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 23: 2116-2125. doi: 10.1038/sj.emboj.7600217
|
| [68] |
Siddall NA, Lin JI, Hime GR, et al. (2009) Myc--what we have learned from flies. Curr Drug Targets 10: 590-601. doi: 10.2174/138945009788680400
|
| [69] |
Cranna N, Quinn L (2009) Impact of steroid hormone signals on Drosophila cell cycle during development. Cell Div 4: 3. doi: 10.1186/1747-1028-4-3
|
| [70] |
Baker NE (2007) Patterning signals and proliferation in Drosophila imaginal discs. Curr Opin Genet Dev 17: 287-293. doi: 10.1016/j.gde.2007.05.005
|
| [71] | Chung HJ, Levens D (2005) c-myc expression: keep the noise down!. Mol Cells 20: 157-166. |
| [72] | Liu J, Levens D (2006) Making myc. Curr Top Microbiol 302: 1-32. |
| [73] | Levens D, Gupta A (2010) Molecular biology. Reliable noise. Science 327: 1088-1089. |
| [74] | Avigan MI, Strober B, Levens D (1990) A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem 265: 18538-18545. |
| [75] | Michelotti GA, Michelotti EF, Pullner A, et al. (1996) Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol Cell Biol 16: 2656-2669. |
| [76] |
Liu J, Akoulitchev S, Weber A, et al. (2001) Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 104: 353-363. doi: 10.1016/S0092-8674(01)00223-9
|
| [77] |
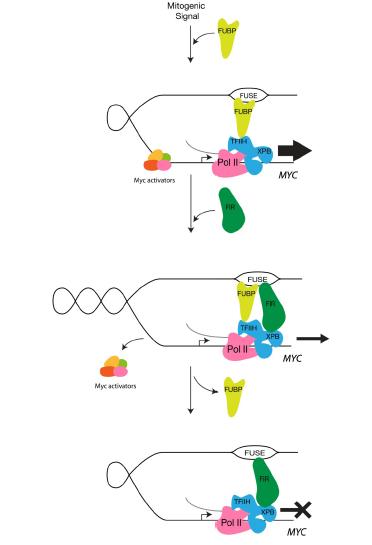
Liu J, Kouzine F, Chung HJ, et al. (2006) The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J 25: 2119-2130. doi: 10.1038/sj.emboj.7601101
|
| [78] |
Matsushita K, Tomonaga T, Shimada H, et al. (2006) An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res 66: 1409-1417. doi: 10.1158/0008-5472.CAN-04-4459
|
| [79] |
Quinn LM, Dickins RA, Coombe M, et al. (2004) Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development 131:1411-1423. doi: 10.1242/dev.01019
|
| [80] |
Mitchell NC, Johanson TM, Cranna NJ, et al. (2010) Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development 137: 2875-2884. doi: 10.1242/dev.049585
|
| [81] | He TC, Sparks AB, Rago C, et al. (1998) Identification of c-MYC as a target of the APC pathway. Science 281: 1509-1512. |
| [82] |
Korinek V, Barker N, Morin PJ, et al. (1997) Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 275: 1784-1787. doi: 10.1126/science.275.5307.1784
|
| [83] |
Morin PJ, Sparks AB, Korinek V, et al. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275: 1787-1790. doi: 10.1126/science.275.5307.1787
|
| [84] |
Korinek V, Barker N, Moerer P, et al. (1998) Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 19: 379-383. doi: 10.1038/1270
|
| [85] |
Pinto D, Gregorieff A, Begthel H, et al. (2003) Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 17: 1709-1713. doi: 10.1101/gad.267103
|
| [86] |
van de Wetering M, Sancho E, Verweij C, et al. (2002) The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241-250. doi: 10.1016/S0092-8674(02)01014-0
|
| [87] |
Muncan V, Sansom OJ, Tertoolen L, et al. (2006) Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol 26: 8418-8426. doi: 10.1128/MCB.00821-06
|
| [88] |
Sansom OJ, Meniel VS, Muncan V, et al. (2007) Myc deletion rescues Apc deficiency in the small intestine. Nature 446: 676-679. doi: 10.1038/nature05674
|
| [89] | Jonsson M, Dejmek J, Bendahl PO, et al. (2002) Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res 62: 409-416. |
| [90] |
Winn RA, Marek L, Han SY, et al. (2005) Restoration of Wnt-7a expression reverses non-small cell lung cancer cellular transformation through frizzled-9-mediated growth inhibition and promotion of cell differentiation. J Biol Chem 280: 19625-19634. doi: 10.1074/jbc.M409392200
|
| [91] |
Kremenevskaja N, von Wasielewski R, Rao AS, et al. (2005) Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 24: 2144-2154. doi: 10.1038/sj.onc.1208370
|
| [92] |
Ross SE, Erickson RL, Gerin I, et al. (2002) Microarray analyses during adipogenesis: understanding the effects of Wnt signaling on adipogenesis and the roles of liver X receptor alpha in adipocyte metabolism. Mol Cell Biol 22: 5989-5999. doi: 10.1128/MCB.22.16.5989-5999.2002
|
| [93] |
Hirabayashi Y, Itoh Y, Tabata H, et al. (2004) The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131: 2791-2801. doi: 10.1242/dev.01165
|
| [94] |
Johnston LA, Sanders AL (2003) Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat Cell Biol 5: 827-833. doi: 10.1038/ncb1041
|
| [95] |
van Es JH, Jay P, Gregorieff A, et al. (2005) Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7: 381-386. doi: 10.1038/ncb1240
|
| [96] |
Fodde R, Brabletz T (2007) Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 19: 150-158. doi: 10.1016/j.ceb.2007.02.007
|
| [97] |
Atlasi Y, Looijenga L, Fodde R (2014) Cancer stem cells, pluripotency, and cellular heterogeneity: a WNTer perspective. Curr Top Dev Biol 107: 373-404. doi: 10.1016/B978-0-12-416022-4.00013-5
|
| [98] |
Sancho E, Batlle E, Clevers H (2004) Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Bi 20: 695-723. doi: 10.1146/annurev.cellbio.20.010403.092805
|
| [99] | Kelleher FC, Fennelly D, Rafferty M (2006) Common critical pathways in embryogenesis and cancer, Acta Oncol 45: 375-388. |
| [100] |
Hopfer O, Zwahlen D, Fey MF, et al. (2005) The Notch pathway in ovarian carcinomas and adenomas. Brit J Cancer 93: 709-718. doi: 10.1038/sj.bjc.6602719
|
| [101] | de Celis JF, Garcia-Bellido A, Bray SJ (1996) Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development 122: 359-369. |
| [102] |
Johnston LA, Edgar BA (1998) Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature 394: 82-84. doi: 10.1038/27925
|
| [103] |
Herranz H, Pérez L, Martín FA, et al. (2008) A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. EMBO J 27: 1633-1645. doi: 10.1038/emboj.2008.84
|
| [104] |
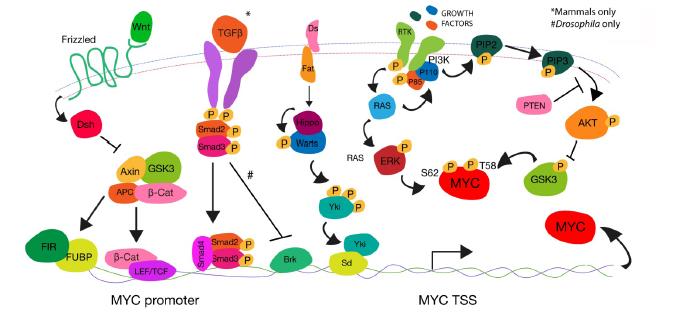
Massague J (2008) TGFbeta in Cancer. Cell 134: 215-230. doi: 10.1016/j.cell.2008.07.001
|
| [105] | Coffey RJ, Bascom CC, Sipes NJ, et al. (1988) Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor beta. Mol Cell Biol 8: 3088-3093. |
| [106] |
Chen CR, Kang Y, Siegel PM, et al. (2002) E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 110: 19-32. doi: 10.1016/S0092-8674(02)00801-2
|
| [107] |
Pennetier D, Oyallon J, Morin-Poulard I, et al. (2012) Size control of the Drosophila hematopoietic niche by bone morphogenetic protein signaling reveals parallels with mammals. PNAS 109: 3389-3394. doi: 10.1073/pnas.1109407109
|
| [108] |
Qu X, Shen L, Zheng Y, et al. (2014) A signal transduction pathway from TGF-β1 to SKP2 via Akt1 and c-Myc and its correlation with progression in human melanoma. J Invest Dermatol 134: 159-167. doi: 10.1038/jid.2013.281
|
| [109] |
Burke R, Basler K (1996) Hedgehog-dependent patterning in the Drosophila eye can occur in the absence of Dpp signaling. Dev Biol 179: 360-368. doi: 10.1006/dbio.1996.0267
|
| [110] | Martin-Castellanos C, Edgar BA (2002) A characterization of the effects of Dpp signaling on cell growth and proliferation in the Drosophila wing. Development 129: 1003-1013. |
| [111] |
Doumpas N, Ruiz-Romero M, Blanco E, et al. (2013) Brk regulates wing disc growth in part via repression of Myc expression. EMBO Rep 14: 261-268. doi: 10.1038/embor.2013.1
|
| [112] |
Lawrence PA, Casal J (2013) The mechanisms of planar cell polarity, growth and the Hippo pathway: some known unknowns. Dev Biol 377: 1-8. doi: 10.1016/j.ydbio.2013.01.030
|
| [113] |
Pan D (2010) The hippo signaling pathway in development and cancer. Dev cell 19: 491-505. doi: 10.1016/j.devcel.2010.09.011
|
| [114] |
Neto-Silva RM, de Beco S, Johnston LA (2010) Evidence for a Growth-Stabilizing Regulatory Feedback Mechanism between Myc and Yorkie, the Drosophila Homolog of Yap. Dev Cell 19: 507-520. doi: 10.1016/j.devcel.2010.09.009
|
| [115] |
Mori M, Triboulet R, Mohseni M, et al. (2014) Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 156: 893-906. doi: 10.1016/j.cell.2013.12.043
|
| [116] |
Pan D (2007) Hippo signaling in organ size control. Genes Dev 21: 886-897. doi: 10.1101/gad.1536007
|
| [117] |
Ziosi M, Baena-López LA, Grifoni D, et al. (2010) dMyc functions downstream of Yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet 6: e1001140. doi: 10.1371/journal.pgen.1001140
|
| [118] |
Xiao W, Wang J, Ou C, et al. (2013) Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem Bioph Res Co 439: 167-172. doi: 10.1016/j.bbrc.2013.08.071
|
| [119] |
Orme MH, Alrubaie S, Bradley GL, et al. (2006) Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat Cell Biol 8: 1298-1302. doi: 10.1038/ncb1493
|
| [120] |
Prober DA, Edgar BA (2002) Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev 16:2286-2299. doi: 10.1101/gad.991102
|
| [121] | Leevers SJ, Weinkove D, MacDougall LK, et al. (1996) The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J 15: 6584-6594. |
| [122] |
Hall DJ, Grewal SS, de la Cruz AF, et al. (2007) Rheb-TOR signaling promotes protein synthesis, but not glucose or amino acid import, in Drosophila. BMC Biol 5: 1. doi: 10.1186/1741-7007-5-1
|
| [123] |
Grewal SS, Evans JR, Edgar BA (2007) Drosophila TIF-IA is required for ribosome synthesis and cell growth and is regulated by the TOR pathway. J Cell Biol 179: 1105-1113. doi: 10.1083/jcb.200709044
|
| [124] |
Grewal SS (2009) Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int J Biochem Cell Biol 41: 1006-1010. doi: 10.1016/j.biocel.2008.10.010
|
| [125] | Li L, Edgar BA, Grewal SS (2010) Nutritional control of gene expression in Drosophila larvae via TOR, Myc and a novel cis-regulatory element. BMC Cell Biol 11: 7. |
| [126] | Edgar BA (2006) How flies get their size: genetics meets physiology. Nat Rev 7: 907-916. |
| [127] |
Britton JS, Lockwood WK, Li L, et al. (2002) Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2: 239-249. doi: 10.1016/S1534-5807(02)00117-X
|
| [128] |
Jiang BH, Liu LZ (2009) PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res 102: 19-65. doi: 10.1016/S0065-230X(09)02002-8
|
| [129] |
Teleman AA, Hietakangas V, Sayadian AC, et al. (2008) Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metab 7: 21-32. doi: 10.1016/j.cmet.2007.11.010
|
| [130] |
Parisi F, Riccardo S, Daniel M, et al. (2011) Drosophila insulin and target of rapamycin (TOR) pathways regulate GSK3 beta activity to control Myc stability and determine Myc expression in vivo. BMC Biol 9: 65. doi: 10.1186/1741-7007-9-65
|
| [131] |
Amcheslavsky A, Ito N, Jiang J, et al. (2011) Tuberous sclerosis complex and Myc coordinate the growth and division of Drosophila intestinal stem cells. J Cell Biol 193: 695-710. doi: 10.1083/jcb.201103018
|
| [132] | Chan JC, Hannan KM, Riddell K, et al. (2011) AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal 4: ra56. |
| [133] | Hann SR, Eisenman RN (1984) Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cellular Biol 4: 2486-2497. |
| [134] |
Gregory MA, Hann SR (2000) c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells. Mol Cellular Biol 20: 2423-2435. doi: 10.1128/MCB.20.7.2423-2435.2000
|
| [135] | Farrell AS, Sears RC (2014) MYC degradation. Cold Spring Harb Perspect Med 4: 3. |
| [136] |
Wang C, Lisanti MP, Liao DJ (2011) Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 10: 57-67. doi: 10.4161/cc.10.1.14449
|
| [137] | Sears R, Nuckolls F, Haura E, et al. (2000) Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability, Genes Dev 14: 2501-2514. |
| [138] | Papas TS, Lautenberger JA (1985) Sequence curiosity in v-myc oncogene. Nature 318: p. 237. |
| [139] |
Gregory MA, Qi Y, Hann SR (2003) Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J Biol Chem 278: 51606-51612. doi: 10.1074/jbc.M310722200
|
| [140] |
Yeh E, Cunningham M, Arnold H, et al. (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6: 308-318. doi: 10.1038/ncb1110
|
| [141] |
Davis MA, Larimore EA, Fissel BM, et al. (2013) The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev 27: 151-156. doi: 10.1101/gad.207720.112
|
| [142] |
Cross DA, Alessi DR, Cohen P, et al. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789. doi: 10.1038/378785a0
|
| [143] |
Welcker M, Orian A, Jin J, et al. (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 101: 9085-9090. doi: 10.1073/pnas.0402770101
|
| [144] |
Galletti M, Riccardo S, Parisi F, et al. (2009) Identification of domains responsible for ubiquitin-dependent degradation of dMyc by glycogen synthase kinase 3beta and casein kinase 1 kinases. Mol Cell Biol 29: 3424-3434. doi: 10.1128/MCB.01535-08
|
| [145] |
Moberg KH, Mukherjee A, Veraksa A, et al. (2004) The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol 14: 965-974. doi: 10.1016/j.cub.2004.04.040
|
| [146] |
Arnold HK, Zhang X, Daniel CJ, et al. (2009) The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J 28: 500-512. doi: 10.1038/emboj.2008.279
|
| [147] |
Johnston LA (2009) Competitive Interactions Between Cells: Death, Growth, and Geography. Science 324: 1679-1682. doi: 10.1126/science.1163862
|
| [148] |
de la Cova C, Abril M, Bellosta P, et al (2004) Drosophila myc regulates organ size by inducing cell competition. Cell 117: 107-116. doi: 10.1016/S0092-8674(04)00214-4
|
| [149] |
Moreno E, Basler K (2004) dMyc transforms cells into super-competitors. Cell 117: 117-129. doi: 10.1016/S0092-8674(04)00262-4
|
| [150] |
Baker NE, Li W. (2008) Cell competition and its possible relation to cancer. Cancer Res 68: 5505-5507. doi: 10.1158/0008-5472.CAN-07-6348
|
| [151] |
Moreno E (2008) Is cell competition relevant to cancer?. Nat Rev 8: 141-147. doi: 10.1038/nrc2252
|
| [152] | Johnston LA (2014) Socializing with MYC: cell competition in development and as a model for premalignant cancer. Cold Spring Harb Perspect Med 4: a014274. |
| [153] |
Senoo-Matsuda N, Johnston LA (2007) Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc Natl Acad Sci U S A 104: 18543-18548. doi: 10.1073/pnas.0709021104
|
| [154] |
Bywater MJ, Pearson RB, McArthur GA, et al. (2013) Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat Rev 13: 299-314. doi: 10.1038/nrc3496
|
Figures(2)

Jue Er Amanda Lee, Linda May Parsons, Leonie M. Quinn. MYC function and regulation in flies: how Drosophila has enlightened MYC cancer biology[J]. AIMS Genetics, 2014, 1(1): 81-98. doi: 10.3934/genet.2014.1.81







 DownLoad:
DownLoad: