Citation: Anna Mainka, Barbara Kozielska. Assessment of the BTEX concentrations and health risk in urban nursery schools in Gliwice, Poland[J]. AIMS Environmental Science, 2016, 3(4): 858-870. doi: 10.3934/environsci.2016.4.858

| [1] |
Ashmore MR, Dimitroulopoulou C (2009) Personal exposure of children to air pollution. Atmos Environ 43: 128-141. doi: 10.1016/j.atmosenv.2008.09.024

|
| [2] |
Son B, Breysse P, Yang W (2003) Volatile organic compounds concentrations in residential indoor and outdoor and its personal exposure in Korea. Environ Int 29: 79-85. doi: 10.1016/S0160-4120(02)00148-4
|
| [3] |
Park KH, Jo WK (2004) Personal volatile organic compound (VOC) exposure of children attending elementary schools adjacent to industrial complex. Atmos Environ 38: 1303-1312. doi: 10.1016/j.atmosenv.2003.11.032
|
| [4] |
Salvi S (2007) Health effects of ambient air pollution in children. Paediatr Respir Rev 8: 275-280. doi: 10.1016/j.prrv.2007.08.008
|
| [5] |
Daisey JM, Angell WJ, Apte MG, et al. (2003) Indoor air quality, ventilation and health symptoms in schools: an analysis of existing information. Indoor Air 13: 53-64. doi: 10.1034/j.1600-0668.2003.00153.x
|
| [6] |
Sofuoglu SC, Aslan G, Inal F, et al. (2011) An assessment of indoor air concentrations and health risks of volatile organic compounds in three primary schools. Int J Hyg Environ Health 214: 36-46. doi: 10.1016/j.ijheh.2010.08.008
|
| [7] |
de Gennaro G, Farella G, Marzocca A, et al. (2013) Indoor and outdoor monitoring of volatile organic compounds in school buildings: Indicators based on health risk assessment to single out critical issues. Int J Environ Res Public Health 10: 6273-6291. doi: 10.3390/ijerph10126273
|
| [8] |
Godwin C, Batterman S (2007) Indoor air quality in Michigan schools. Indoor Air 17: 109-121. doi: 10.1111/j.1600-0668.2006.00459.x
|
| [9] |
Rizk M, Verriele M, Dusanter S, et al. (2016) Fast sorption measurements of volatile organic compounds on building materials: Part 1 - Methodology developed for field applications. Data Br 6: 953-958. doi: 10.1016/j.dib.2016.01.011
|
| [10] | ASHRAE (1999) Standard 62-1999 Ventilation for Acceptable Indoor Air Quality. |
| [11] | IARC (2015) International Agency for Research on Cancer: Monographs on the Evaluation of Carcinogenic Risks to Humans. Available from: http: //monographs.iarc.fr/ENG/Classification/ index.php |
| [12] | Mendell MJ, Heath GA (2005) Do indoor pollutants and thermal conditions in schools influence student performance? A critical review of the literature. Indoor Air 15: 27-52. |
| [13] |
Santamouris M, Synnefa A, Asssimakopoulos M, et al. (2008) Experimental investigation of the air flow and indoor carbon dioxide concentration in classrooms with intermittent natural ventilation. Energy Build 40: 1833-1843. doi: 10.1016/j.enbuild.2008.04.002
|
| [14] |
Yoon C, Lee K, Park D (2011) Indoor air quality differences between urban and rural preschools in Korea. Environ Sci Pollut Res 18: 333-345. doi: 10.1007/s11356-010-0377-0
|
| [15] |
Branco PTBS, Alvim-Ferraz MCM, Martins FG, et al. (2014) Indoor air quality in urban nurseries at Porto city: Particulate matter assessment. Atmos Environ 84: 133-143. doi: 10.1016/j.atmosenv.2013.11.035
|
| [16] |
Mainka A, Brągoszewska E, Kozielska B, et al. (2015) Indoor air quality in urban nursery schools in Gliwice, Poland: Analysis of the case study. Atmos Pollut Res 6: 1098-1104. doi: 10.1016/j.apr.2015.06.007
|
| [17] | Mainka A, Zajusz-Zubek E, Kozielska B, et al. (2015) Badanie zanieczyszczeń powietrza oddziałujących na dzieci w przedszkolu miejskim zlokalizowanym przy drodze o dużym natężeniu ruchu. Inżynieria i Ochr Środowiska 18: 119-133. |
| [18] | Mainka A, Zajusz-Zubek E, Kozielska B, et al. (2014) Badanie zanieczyszczeń powietrza oddziałujących na dzieci w wybranym przedszkolu miejskim. In: Konieczyński J (ed) Ochr powietrza w Teor i Prakt Instytut Podstaw Inżynierii Środowiska Polskiej Akademii Nauk, Zabrze, 1: 115-128. |
| [19] | Kozielska B (2013) Concentration of benzene and its alkyl derivatives in Gliwice air. Arch Environ Prot Waste Manag Environ Prot 15: 81-88. |
| [20] |
Brągoszewska E, Mainka A, Pastuszka JS (2016) Bacterial aerosols in an urban nursery school in Gliwice, Poland: a case study. Aerobiologia 32: 469-480. doi: 10.1007/s10453-015-9419-x
|
| [21] | SKC (2012) VOC method update SKC appendices to EPA method TO-17 determination of volatile organic compounds (VOCs) in ambient or indoor air. |
| [22] | Dz.U. (2012) (Journal of Laws) Regulation of the Minister of Environment of 13 September 2012, No. 1031 - The levels of certain substances in the ambient air. |
| [23] |
Demirel G, Özden Ö, Döǧeroǧlu T, et al. (2014) Personal exposure of primary school children to BTEX, NO2 and ozone in Eskişehir, Turkey: Relationship with indoor/outdoor concentrations and risk assessment. Sci Total Environ 473-474: 537-548. doi: 10.1016/j.scitotenv.2013.12.034
|
| [24] |
Marć M, Zabiegała B, Simeonov V, et al. (2014) The Relationships Between BTEX, NOx, and O3 Concentrations in Urban Air in Gdansk and Gdynia, Poland. CLEAN Soil Air Water 42: 1326-1336. doi: 10.1002/clen.201300188
|
| [25] | Rogula-Kozłowska W, Pastuszka JS, Talik E (2008) Influence of vehicular traffic on concentration and particle surface composition of PM10 and PM2.5 in Zabrze, Poland. Polish J Environ Stud 17: 539-548. |
| [26] |
Jung KH, Artigas F, Shin JY (2011) Personal, indoor, and outdoor exposure to VOCs in the immediate vicinity of a local airport. Environ Monit Assess 173: 555-567. doi: 10.1007/s10661-010-1404-9
|
| [27] |
Buczyńska AJ, Krata A, Stranger M, et al. (2009) Atmospheric BTEX-concentrations in an area with intensive street traffic. Atmos Environ 43: 311-318. doi: 10.1016/j.atmosenv.2008.09.071
|
| [28] |
Delgado-Saborit JM, Aquilina NJ, Meddings C, et al. (2011) Relationship of personal exposure to volatile organic compounds to home, work and fixed site outdoor concentrations. Sci Total Environ 409: 478-488. doi: 10.1016/j.scitotenv.2010.10.014
|
| [29] | EPA U (1999) Environmental Technology Protocol Verification Report Emissions of VOCs and Aldehydes from Commercial Furniture. |
| [30] |
Kotzias D (2005) Indoor air and human exposure assessment - Needs and approaches. Exp Toxicol Pathol 57: 5-7. doi: 10.1016/j.etp.2005.05.002
|
| [31] | ATSDR (2015) Agency for Toxic Substances and Diseases Registry. Available from: http://www.atsdr.cdc.gov/toxfaq.html. |
| [32] | Guo H, Lee SC, Li WM, et al. (2003) Source characterization of BTEX in indoor microenvironments in Hong Kong. Atmos Environ 37: 73-82. |
| [33] |
Hoque RR, Khillare PS, Agarwal T, et al. (2008) Spatial and temporal variation of BTEX in the urban atmosphere of Delhi, India. Sci Total Environ 392: 30-40. doi: 10.1016/j.scitotenv.2007.08.036
|
| [34] | ASHRAE (2009) ASHRAE Handbook - Fundamentals (SI Edition). American Society of Heating, Refrigerating and Air-Conditioning Engineers, Inc. |
| [35] | WHO (2010) WHO guidelines for indoor air quality: selected pollutants. |
| [36] |
Kim KH, Jahan SA, Kabir E (2011) A review of diseases associated with household air pollution due to the use of biomass fuels. J Hazard Mater 192: 425-431. doi: 10.1016/j.jhazmat.2011.05.087
|
| [37] |
Wu XM, Fan ZT, Zhu X, et al. (2012) Exposures to volatile organic compounds (VOCs) and associated health risks of socio-economically disadvantaged population in a “hot spot” in Camden, New Jersey. Atmos Environ 57: 72-79. doi: 10.1016/j.atmosenv.2012.04.029
|
| [38] |
Hinwood AL, Rodriguez C, Runnion T, et al. (2007) Risk factors for increased BTEX exposure in four Australian cities. Chemosphere 66: 533-541. doi: 10.1016/j.chemosphere.2006.05.040
|
| [39] | U.S. EPA (2002) Child-Specific Exposure Factors Handbook. EPA/600/R-06/096F. |
| [40] | Johnson-Restrepo B, Kannan K (2009) An assessment of sources and pathways of human exposure to polybrominated diphenyl ethers in the United States. Chemosphere 76: 542-548. |
| [41] | U.S. EPA (2004) Risk Assessment: “Supplemental Guidance for Dermal Risk Assessment”, Part E of Risk Assessment Guidance for Superfund, Human Health Evaluation Manual (Volume I), August 16, 2004. |
| [42] | Ott WR, Steinemann AC, Wallace LA (2006) Exposure Analysis, CRC Press, London. |
| [43] |
Guo H, Lee SC, Chan LY, et al. (2004) Risk assessment of exposure to volatile organic compounds in different indoor environments. Environ Res 94: 57-66. doi: 10.1016/S0013-9351(03)00035-5
|
| [44] |
Li H, Qian X, Hu W, et al. (2013) Chemical speciation and human health risk of trace metals in urban street dusts from a metropolitan city, Nanjing, SE China. Sci Total Environ 456-457: 212-221. doi: 10.1016/j.scitotenv.2013.03.094
|
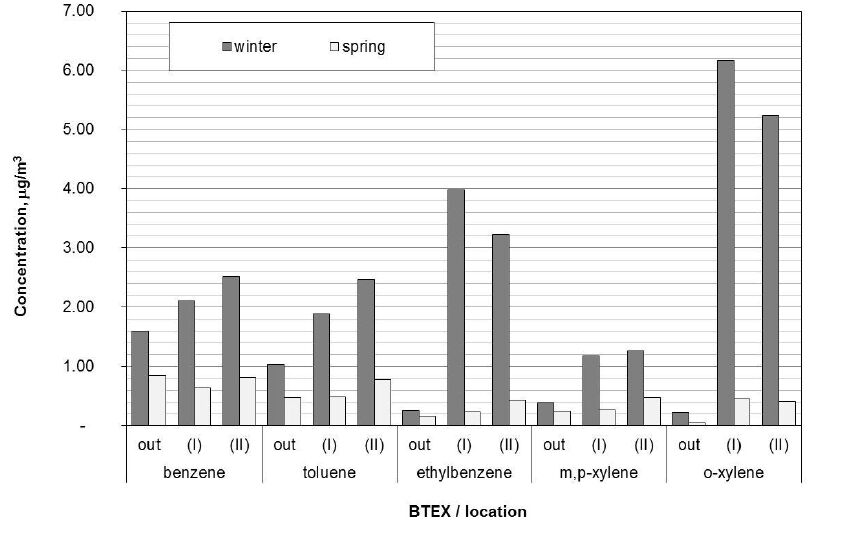
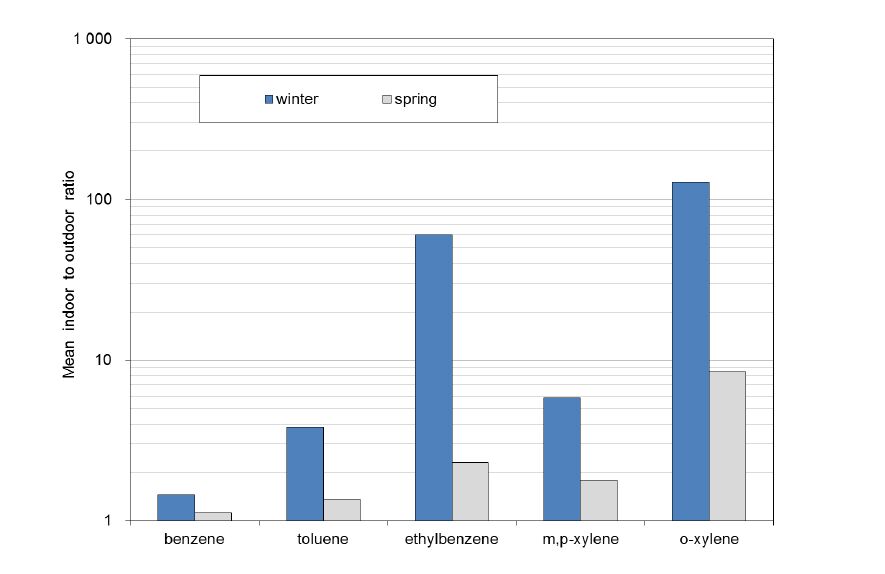
Figures(2) / Tables(2)

Anna Mainka, Barbara Kozielska. Assessment of the BTEX concentrations and health risk in urban nursery schools in Gliwice, Poland[J]. AIMS Environmental Science, 2016, 3(4): 858-870. doi: 10.3934/environsci.2016.4.858







 DownLoad:
DownLoad: