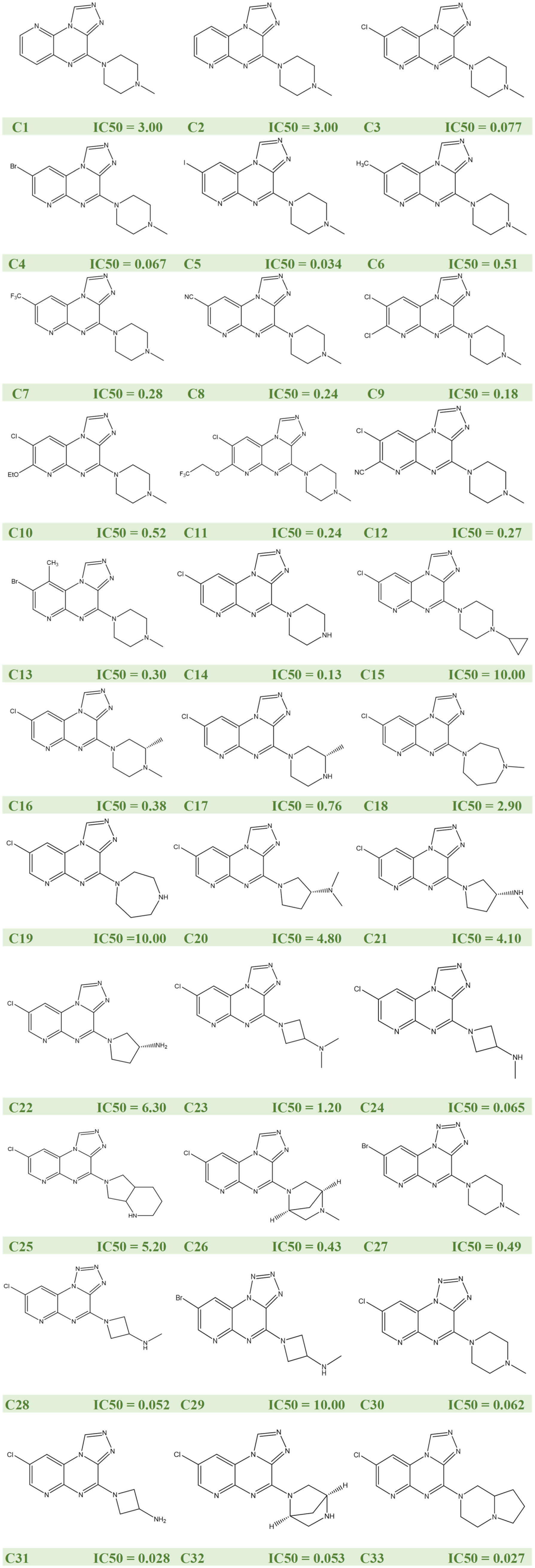
Atopic dermatitis (AD) is a prevalent inflammatory skin condition, primarily characterized by intense pruritus and chronic inflammation. Current therapeutic options targeting the histamine H4 receptor (H4R) have shown limited efficacy in addressing both pruritus and inflammation comprehensively. This study investigates pyridopyrazine derivatives as potential H4R antagonists with a focus on their suitability for AD treatment. To evaluate these compounds, we applied quantitative structure–activity relationship (QSAR) models and molecular docking techniques. A set of 33 pyridopyrazine derivatives was analyzed using principal component regression (PCR), multiple linear regression (MLR), and partial least squares (PLS) methodologies. Molecular descriptors were computed, and collinearity among descriptors was assessed through principal component analysis (PCA). Model performance was evaluated using the root mean square error (RMSE) and coefficient of determination (R2) values, providing insight into predictive accuracy. The PCR model emerged with strong predictive capabilities, showing an RMSE of 1.017 and an R2 of 0.897. Furthermore, molecular docking results indicated potent binding interactions with H4R, primarily through hydrophobic and hydrogen-bonding interactions. Notably, compound C11 demonstrated the highest binding affinity, underscoring its potential as a valuable candidate for anti-inflammatory development. In conclusion, pyridopyrazine derivatives, particularly compound C11, exhibit promising anti-inflammatory properties with specific binding efficacy to H4R, suggesting potential for advancing AD treatment options.
Citation: Mohamed El Yaqoubi, Mouad Lahyaoui, Yousra Seqqat, Taoufiq Saffaj, Bouchaib Ihssane, Nabil Saffaj, Rachid Mamouni, Fouad Ouazzani Chahdi, Fahad M Alshabrmi, Alaa Abdulaziz Alnahari, Saad M. Howladar, Ammar AL-Farga, Youssef Kandri Rodi. Pyridopyrazine derivatives as highly selective histamine H4 receptor antagonist for the treatment of atopic dermatitis: QSAR modeling and molecular docking studies[J]. AIMS Allergy and Immunology, 2024, 8(4): 303-323. doi: 10.3934/Allergy.2024019
Atopic dermatitis (AD) is a prevalent inflammatory skin condition, primarily characterized by intense pruritus and chronic inflammation. Current therapeutic options targeting the histamine H4 receptor (H4R) have shown limited efficacy in addressing both pruritus and inflammation comprehensively. This study investigates pyridopyrazine derivatives as potential H4R antagonists with a focus on their suitability for AD treatment. To evaluate these compounds, we applied quantitative structure–activity relationship (QSAR) models and molecular docking techniques. A set of 33 pyridopyrazine derivatives was analyzed using principal component regression (PCR), multiple linear regression (MLR), and partial least squares (PLS) methodologies. Molecular descriptors were computed, and collinearity among descriptors was assessed through principal component analysis (PCA). Model performance was evaluated using the root mean square error (RMSE) and coefficient of determination (R2) values, providing insight into predictive accuracy. The PCR model emerged with strong predictive capabilities, showing an RMSE of 1.017 and an R2 of 0.897. Furthermore, molecular docking results indicated potent binding interactions with H4R, primarily through hydrophobic and hydrogen-bonding interactions. Notably, compound C11 demonstrated the highest binding affinity, underscoring its potential as a valuable candidate for anti-inflammatory development. In conclusion, pyridopyrazine derivatives, particularly compound C11, exhibit promising anti-inflammatory properties with specific binding efficacy to H4R, suggesting potential for advancing AD treatment options.
atopic dermatitis
accessible surface area (positive charge)
accessible surface area (negative charge)
accessible surface area (polar)
atom site violation descriptor
histamine H4 receptor
inhibitory concentration 50%
immunoglobulin E
multiple linear regression
negative logarithm of IC50
principal component analysis
principal component regression
partial least squares
quantitative structure–activity relationship
coefficient of determination
root mean square error
resonance synthetic descriptor
T-helper (e.g., TH1, TH17, TH22 cytokines)
Vertex Adjacency Equality Index
molecular surface properties descriptor
VSURF coefficient of polarizability
VSURF hydrophobic constant (Cavity 4)
VSURF Hydrophilic–Lipophilic Index 1
VSURF Hydrophilic–Lipophilic Index 2
Wiener Polarity Index (descriptor of molecular branching)
software for statistical analysis
Zagreb Index Descriptor (topological descriptor)

| [1] |
Jolliffe IT, Cadima J (2016) Principal component analysis: A review and recent developments. Philos Trans R Soc A 374: 20150202. https://doi.org/10.1098/rsta.2015.0202 
|
| [2] |
Kim J, Kim BE, Leung DYM (2019) Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma Proc 40: 84-92. https://doi.org/10.2500/aap.2019.40.4202
|
| [3] |
David Boothe W, Tarbox JA, Tarbox MB (2017) Atopic dermatitis: Pathophysiology. Management of Atopic Dermatitis . Cham: Springer International Publishing 21-37. https://doi.org/10.1007/978-3-319-64804-0_3
|
| [4] |
Sroka-Tomaszewska J, Trzeciak M (2021) Molecular mechanisms of atopic dermatitis pathogenesis. Int J Mol Sci 22: 4130. https://doi.org/10.3390/ijms22084130
|
| [5] |
Maintz L, Bieber T, Simpson HD, et al. (2022) From skin barrier dysfunction to systemic impact of atopic dermatitis: Implications for a precision approach in dermocosmetics and medicine. J Pers Med 12: 893. https://doi.org/10.3390/jpm12060893
|
| [6] | Santamaria-Babí LF (2022) Atopic dermatitis pathogenesis: Lessons from immunology. Dermatol Pract Concept 12. https://doi.org/10.5826/dpc.1201a152 |
| [7] |
Guttman-Yassky E, Dhingra N, Leung DYM (2013) New era of biologic therapeutics in atopic dermatitis. Expert Opin Biol Ther 13: 549-561. https://doi.org/10.1517/14712598.2013.758708
|
| [8] |
Mitchell PM, Al-Janabi H, Richardson J, et al. (2015) The relative impacts of disease on health status and capability wellbeing: A multi-country study. PLoS ONE 10: e0143590. https://doi.org/10.1371/journal.pone.0143590
|
| [9] |
Brunner PM, Guttman-Yassky E, Leung DYM (2017) The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol 139: S65-S76. https://doi.org/10.1016/j.jaci.2017.01.011
|
| [10] |
Mathew B, Srivastava S, Ross LJ, et al. (2011) Novel pyridopyrazine and pyrimidothiazine derivatives as FtsZ inhibitors. Bioorg Med Chem 19: 7120-7128. https://doi.org/10.1016/j.bmc.2011.09.062
|
| [11] |
Lobo V, Patil A, Phatak A, et al. (2010) Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev 4: 118-126. https://doi.org/10.4103/0973-7847.70902
|
| [12] | Patra AK (2012) An overview of antimicrobial properties of different classes of phytochemicals. Dietary Phytochemicals and Microbes . Cham: Springer 1-32. https://doi.org/10.1007/978-94-007-3926-0_1 |
| [13] | Pizzino G, Irrera N, Cucinotta M, et al. (2017) Oxidative stress: Harms and benefits for human health. Oxid Med Cell Longevity . https://doi.org/10.1155/2017/8416763 |
| [14] |
Pettersson M, Johnson DS, Humphrey JM, et al. (2015) Design of pyridopyrazine-1,6-dione γ-secretase modulators that align potency, MDR efflux ratio, and metabolic stability. ACS Med Chem Lett 6: 596-601. https://doi.org/10.1021/acsmedchemlett.5b00070
|
| [15] |
Pettersson M, Johnson DS, Rankic DA, et al. (2017) Discovery of cyclopropylchromane-derived pyridopyrazine-1,6-dione γ-secretase modulators with robust central efficacy. Med Chem Comm 8: 730-743. https://doi.org/10.1039/c6md00406g
|
| [16] |
Kékesi L, Sipos A, Németh G, et al. (2013) Synthesis and biological evaluation of novel pyrido [2,3-b] pyrazines inhibiting both erlotinib-sensitive and erlotinib-resistant cell lines. Bioorg Med Chem Lett 23: 6152-6155. https://doi.org/10.1016/j.bmcl.2013.09.005
|
| [17] | Sahu R, Shah K, Gautam Y, et al. (2023) Pyrazine moiety: Recent developments in cancer treatment. Curr Org Chem 27: 6152-6155. http://dx.doi.org/10.2174/1385272827666230816105317 |
| [18] |
Niaz L, Saddique FA, Aslam S, et al. (2020) Recent synthetic methodologies for pyridopyrazines: An update. Synth Commun 50: 2755-2786. https://doi.org/10.1080/00397911.2020.1786123
|
| [19] |
Mahmud AW, Shallangwa GA, Uzairu A (2020) QSAR and molecular docking studies of 1,3-dioxoisoindoline-4-aminoquinolines as potent antiplasmodium hybrid compounds. Heliyon 6: e03449. https://doi.org/10.1016/j.heliyon.2020.e03449
|
| [20] |
Rosell-Hidalgo A, Young L, Moore AL, et al. (2021) QSAR and molecular docking for the search of AOX inhibitors: A rational drug discovery approach. J Comput-Aided Molr Des 35: 245-260. https://doi.org/10.1007/s10822-020-00360-8
|
| [21] |
Daoui O, Elkhattabi S, Chtita S, et al. (2021) QSAR, molecular docking and ADMET properties in silico studies of novel 4,5,6,7-tetrahydrobenzo[D]-thiazol-2-Yl derivatives derived from dimedone as potent anti-tumor agents through inhibition of C-Met receptor tyrosine kinase. Heliyon 7: e07463. https://doi.org/10.1016/j.heliyon.2021.e07463
|
| [22] |
Liu J, Li Y, Zhang HX, et al. (2012) Studies of H4R antagonists using 3D-QSAR, molecular docking and molecular dynamics. J Mol Model 18: 991-1001. https://doi.org/10.1007/s00894-011-1137-x
|
| [23] |
Mehta P, Miszta P, Filipek S (2021) Molecular modeling of histamine receptors—recent advances in drug discovery. Molecules 26: 1778. https://doi.org/10.3390/molecules26061778
|
| [24] |
Lahyaoui M, El-Idrissi H, Saffaj T, et al. (2023) QSAR modeling, molecular docking and molecular dynamic simulation of phosphorus-substituted quinoline derivatives as topoisomerase I inhibitors. Arabian J Chem 16: 104783. https://doi.org/10.1016/j.arabjc.2023.104783
|
| [25] |
Lahyaoui M, Diane A, El-Idrissi, et al. (2023) QSAR modeling and molecular docking studies of 2-oxo-1, 2-dihydroquinoline-4- carboxylic acid derivatives as p-glycoprotein inhibitors for combating cancer multidrug resistance. Heliyon 9: e13020. https://doi.org/10.1016/j.heliyon.2023.e13020
|
| [26] |
Li J, Luo D, Wen T, et al. (2021) Representative feature selection of molecular descriptors in QSAR modeling. J Mol Struct 1244: 131249. https://doi.org/10.1016/j.molstruc.2021.131249
|
| [27] |
Frimayanti N, Yam ML, Lee HB, et al. (2011) Validation of quantitative structure-activity relationship (QSAR) model for photosensitizer activity prediction. Int Mol Sci 12: 8626-8644. https://doi.org/10.3390/ijms12128626
|
| [28] |
Roy K, Ambure P, Kar S (2018) How precise are our quantitative structure-activity relationship derived predictions for new query chemicals?. ACS Omega 3: 11392-11406. https://doi.org/10.1021/acsomega.8b01647
|
| [29] |
Khamouli S, Belaidi S, Bakhouch M, et al. (2022) QSAR modeling, molecular docking, ADMET prediction and molecular dynamics simulations of some 6-arylquinazolin-4-amine derivatives as DYRK1A inhibitors. J Mol Struct 1258: 132659. https://doi.org/10.1016/j.molstruc.2022.132659
|
| [30] |
Li J, Fu A, Zhang L (2019) An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip Sci: Computa Life Sci 11: 320-328. https://doi.org/10.1007/s12539-019-00327-w
|
| [31] |
Abdullahi M, Shallangwa GA, Uzairu A (2020) In silico QSAR and molecular docking simulation of some novel aryl sulfonamide derivatives as inhibitors of H5N1 influenza a virus subtype. Beni-Suef Univ J Basic Appl Sci 9: 2. https://doi.org/10.1186/s43088-019-0023-y
|
| [32] |
El Fadili M, Er-Rajy M, Kara M, et al. (2022) QSAR, ADMET in silico pharmacokinetics, molecular docking and molecular dynamics studies of novel bicyclo (aryl methyl) benzamides as potent glyt1 inhibitors for the treatment of schizophrenia. Pharmaceuticals 15: 670. https://doi.org/10.3390/ph15060670
|
| [33] |
Lahyaoui M, Haoudi A, Kartah BE, et al. (2023) Crystal structure, Hirshfeld surface analysis, intermolecular interaction energies, energy frameworks and DFT calculations of 4-amino-1-(prop-2-yn-1-yl)pyrimidin-2(1H)-one. Acta Crystallogr Sec E: Crystallogr Commun 79: 1183-1189. https://doi.org/10.1107/S2056989023009933
|
| [34] |
Stoean B, Rugina D, Focsan M, et al. (2021) Novel (Phenothiazinyl)vinyl-pyridinium dyes and their potential applications as cellular staining agents. Int J Mol Sci 22: 2985. https://doi.org/10.3390/ijms22062985
|
| [35] |
Ko K, Kim HJ, Ho PS, et al. (2018) Discovery of a novel highly selective histamine h4 receptor antagonist for the treatment of atopic dermatitis. J Med Chem 61: 2949-2961. https://doi.org/10.1021/acs.jmedchem.7b01855
|
| [36] |
Danishuddin, Khan AU (2016) Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discovery Today 21: 1291-1302. https://doi.org/10.1016/j.drudis.2016.06.013
|
| [37] | (1987) Statitcf SoftwareTechnical Institute of Cereals and Fodder. Paris, France: . |
| [38] | Hmamouchi R, Larif M, Adad A, et al. (2014) Structure activity and prediction of biological activities of compound (2-methyl-6-phenylethynylpyridine) derivatives relationships rely on electronic and topological descriptors. J Comput Methods Mol Des 4: 61-71. |
| [39] |
Wold S, Esbensen K, Geladi P (1987) Principal component analysis. Chemom Intell Lab Sys 2: 37-52. https://doi.org/10.1016/0169-7439(87)80084-9
|
| [40] |
David CC, Jacobs DJ (2014) Principal component analysis: A method for determining the essential dynamics of proteins. Protein Dynamics . Cham: Springer 193-226. https://doi.org/10.1007/978-1-62703-658-0_11
|
| [41] |
Golbraikh A, Tropsha A (2002) Beware of q2!. J Mol Graphics Modell 20: 269-276. https://doi.org/10.1016/S1093-3263(01)00123-1
|
| [42] |
Chtita S, Belhassan A, Bakhouch M, et al. (2021) QSAR study of unsymmetrical aromatic disulfides as potent avian SARS-CoV main protease inhibitors using quantum chemical descriptors and statistical methods. Chemom Intell Lab Syst 210: 104266. https://doi.org/10.1016/j.chemolab.2021.104266
|
| [43] |
Lahyaoui M, Filali M, Sghyar R, et al. (2024) Development of novel antibiotics derived from pyridazine: Synthesis, spectroscopic characterization, in vitro antimicrobial activity and molecular docking studies. Results Chem 10: 101699. https://doi.org/10.1016/j.rechem.2024.101699
|
 allergy-08-04-019-s001.pdf allergy-08-04-019-s001.pdf |
 |









Figures(10) / Tables(5)

Mohamed El Yaqoubi, Mouad Lahyaoui, Yousra Seqqat, Taoufiq Saffaj, Bouchaib Ihssane, Nabil Saffaj, Rachid Mamouni, Fouad Ouazzani Chahdi, Fahad M Alshabrmi, Alaa Abdulaziz Alnahari, Saad M. Howladar, Ammar AL-Farga, Youssef Kandri Rodi. Pyridopyrazine derivatives as highly selective histamine H4 receptor antagonist for the treatment of atopic dermatitis: QSAR modeling and molecular docking studies[J]. AIMS Allergy and Immunology, 2024, 8(4): 303-323. doi: 10.3934/Allergy.2024019







 DownLoad:
DownLoad: