Citation: Yu Sato, Hiroyuki Jinnouchi, Atsushi Sakamoto, Anne Cornelissen, Masayuki Mori, Rika Kawakami, Kenji Kawai, Renu Virmani, Aloke V. Finn. Calcification in human vessels and valves: from pathological point of view[J]. AIMS Molecular Science, 2020, 7(3): 183-210. doi: 10.3934/molsci.2020009

| [1] |
Torii S, Jinnouchi H, Sakamoto A, et al. (2020) Vascular responses to coronary calcification following implantation of newer-generation drug-eluting stents in humans: impact on healing. Eur Heart J 41: 786-796. doi: 10.1093/eurheartj/ehz850

|
| [2] |
Watanabe Y, Lefèvre T, Bouvier E, et al. (2015) Prognostic value of aortic root calcification volume on clinical outcomes after transcatheter balloon-expandable aortic valve implantation. Catheter Cardiovasc Interv 86: 1105-1113. doi: 10.1002/ccd.25986
|
| [3] |
Iung B, Vahanian A (2011) Epidemiology of valvular heart disease in the adult. Nat Rev Cardiol 8: 162-172. doi: 10.1038/nrcardio.2010.202
|
| [4] |
Bonow RO, Leon MB, Doshi D, et al. (2016) Management strategies and future challenges for aortic valve disease. Lancet 387: 1312-1323. doi: 10.1016/S0140-6736(16)00586-9
|
| [5] |
Nakahara T, Dweck MR, Narula N, et al. (2017) Coronary Artery Calcification: From Mechanism to Molecular Imaging. JACC Cardiovasc Imaging 10: 582-593. doi: 10.1016/j.jcmg.2017.03.005
|
| [6] |
Otsuka F, Sakakura K, Yahagi K, et al. (2014) Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol 34: 724-736. doi: 10.1161/ATVBAHA.113.302642
|
| [7] |
Stary HC, Blankenhorn DH, Chandler AB, et al. (1992) A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 85: 391-405. doi: 10.1161/01.CIR.85.1.391
|
| [8] |
Orlandi A (2015) The contribution of resident vascular stem cells to arterial pathology. Int J Stem Cells 8: 9-17. doi: 10.15283/ijsc.2015.8.1.9
|
| [9] |
Harper E, Forde H, Davenport C, et al. (2016) Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol 82: 30-40. doi: 10.1016/j.vph.2016.02.003
|
| [10] |
Fadini GP, Rattazzi M, Matsumoto T, et al. (2012) Emerging role of circulating calcifying cells in the bone-vascular axis. Circulation 125: 2772-2781. doi: 10.1161/CIRCULATIONAHA.112.090860
|
| [11] |
Burke AP, Weber DK, Kolodgie FD, et al. (2001) Pathophysiology of calcium deposition in coronary arteries. Herz 26: 239-244. doi: 10.1007/PL00002026
|
| [12] |
Mori H, Torii S, Kutyna M, et al. (2018) Coronary Artery Calcification and its Progression: What Does it Really Mean? JACC Cardiovasc Imaging 11: 127-142. doi: 10.1016/j.jcmg.2017.10.012
|
| [13] |
Sangiorgi G, Rumberger JA, Severson A, et al. (1998) Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol 31: 126-133. doi: 10.1016/S0735-1097(97)00443-9
|
| [14] |
Glagov S, Weisenberg E, Zarins CK, et al. (1987) Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 316: 1371-1375. doi: 10.1056/NEJM198705283162204
|
| [15] |
Burke AP, Virmani R, Galis Z, et al. (2003) 34th Bethesda Conference: Task force #2--What is the pathologic basis for new atherosclerosis imaging techniques? J Am Coll Cardiol 41: 1874-1886. doi: 10.1016/S0735-1097(03)00359-0
|
| [16] |
Williams JK, Adams MR, Klopfenstein HS (1990) Estrogen modulates responses of atherosclerotic coronary arteries. Circulation 81: 1680-1687. doi: 10.1161/01.CIR.81.5.1680
|
| [17] |
Burke AP, Taylor A, Farb A, et al. (2000) Coronary calcification: insights from sudden coronary death victims. Z Kardiol 2: 49-53. doi: 10.1007/s003920070099
|
| [18] |
Burke AP, Farb A, Malcom G, et al. (2001) Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am Heart J 141: S58-62. doi: 10.1067/mhj.2001.109946
|
| [19] |
Manson JE, Allison MA, Rossouw JE, et al. (2007) Estrogen therapy and coronary-artery calcification. N Engl J Med 356: 2591-2602. doi: 10.1056/NEJMoa071513
|
| [20] |
Bild DE, Detrano R, Peterson D, et al. (2005) Ethnic differences in coronary calcification: the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation 111: 1313-1320. doi: 10.1161/01.CIR.0000157730.94423.4B
|
| [21] | Burke A, Farb A, Kutys R, et al. (2002) Atherosclerotic coronary plaques in African Americans are less likely to calcify than coronary plaques in Caucasian Americans. Circulation 106: 481-481. |
| [22] | Lee TC, O'Malley PG, Feuerstein I, et al. (2003) The prevalence and severity of coronary artery calcification on coronary artery computed tomography in black and white subjects. J Am Coll Cardiol 41: 39-44. |
| [23] |
Loria CM, Liu K, Lewis CE, et al. (2007) Early adult risk factor levels and subsequent coronary artery calcification: the CARDIA Study. J Am Coll Cardiol 49: 2013-2020. doi: 10.1016/j.jacc.2007.03.009
|
| [24] |
Kiel DP, Kauppila LI, Cupples LA, et al. (2001) Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham Heart Study. Calcif Tissue Int 68: 271-276. doi: 10.1007/BF02390833
|
| [25] |
Mauriello A, Servadei F, Zoccai GB, et al. (2013) Coronary calcification identifies the vulnerable patient rather than the vulnerable Plaque. Atherosclerosis 229: 124-129. doi: 10.1016/j.atherosclerosis.2013.03.010
|
| [26] |
Huang CC, Lloyd-Jones DM, Guo X, et al. (2011) Gene expression variation between African Americans and whites is associated with coronary artery calcification: the multiethnic study of atherosclerosis. Physiol Genomics 43: 836-843. doi: 10.1152/physiolgenomics.00243.2010
|
| [27] |
Fornage M, Boerwinkle E, Doris PA, et al. (2004) Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 109: 335-339. doi: 10.1161/01.CIR.0000109487.46725.02
|
| [28] |
Rumberger JA, Simons DB, Fitzpatrick LA, et al. (1995) Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. A histopathologic correlative study. Circulation 92: 2157-2162. doi: 10.1161/01.CIR.92.8.2157
|
| [29] |
Raggi P, Shaw LJ, Berman DS, et al. (2004) Prognostic value of coronary artery calcium screening in subjects with and without diabetes. J Am Coll Cardiol 43: 1663-1669. doi: 10.1016/j.jacc.2003.09.068
|
| [30] |
Carson AP, Steffes MW, Carr JJ, et al. (2015) Hemoglobin a1c and the progression of coronary artery calcification among adults without diabetes. Diabetes care 38: 66-71. doi: 10.2337/dc14-0360
|
| [31] |
Burke AP, Kolodgie FD, Farb A, et al. (2001) Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation 103: 934-940. doi: 10.1161/01.CIR.103.7.934
|
| [32] |
Burke AP, Kolodgie FD, Zieske A, et al. (2004) Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler Thromb Vasc Biol 24: 1266-1271. doi: 10.1161/01.ATV.0000131783.74034.97
|
| [33] |
Goodman WG, Goldin J, Kuizon BD, et al. (2000) Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342: 1478-1483. doi: 10.1056/NEJM200005183422003
|
| [34] |
Sigrist M, Bungay P, Taal MW, et al. (2006) Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol Dial Transplant 21: 707-714. doi: 10.1093/ndt/gfi236
|
| [35] |
Baber U, de Lemos JA, Khera A, et al. (2008) Non-traditional risk factors predict coronary calcification in chronic kidney disease in a population-based cohort. Kidney Int 73: 615-621. doi: 10.1038/sj.ki.5002716
|
| [36] |
Block GA, Raggi P, Bellasi A, et al. (2007) Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int 71: 438-441. doi: 10.1038/sj.ki.5002059
|
| [37] |
Raggi P, Boulay A, Chasan-Taber S, et al. (2002) Cardiac calcification in adult hemodialysis patients. A link between end-stage renal disease and cardiovascular disease? J Am Coll Cardiol 39: 695-701. doi: 10.1016/S0735-1097(01)01781-8
|
| [38] |
Oh J, Wunsch R, Turzer M, et al. (2002) Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 106: 100-105. doi: 10.1161/01.CIR.0000020222.63035.C0
|
| [39] |
Chertow GM, Raggi P, Chasan-Taber S, et al. (2004) Determinants of progressive vascular calcification in haemodialysis patients. Nephrol Dial Transplant 19: 1489-1496. doi: 10.1093/ndt/gfh125
|
| [40] |
Chertow GM, Burke SK, Raggi P (2002) Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 62: 245-252. doi: 10.1046/j.1523-1755.2002.00434.x
|
| [41] |
Guerin AP, London GM, Marchais SJ, et al. (2000) Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant 15: 1014-1021. doi: 10.1093/ndt/15.7.1014
|
| [42] |
Adeney KL, Siscovick DS, Ix JH, et al. (2009) Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 20: 381-387. doi: 10.1681/ASN.2008040349
|
| [43] |
Kestenbaum B, Sampson JN, Rudser KD, et al. (2005) Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 16: 520-528. doi: 10.1681/ASN.2004070602
|
| [44] |
Dhingra R, Sullivan LM, Fox CS, et al. (2007) Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 167: 879-885. doi: 10.1001/archinte.167.9.879
|
| [45] |
Palmer SC, Gardner S, Tonelli M, et al. (2016) Phosphate-Binding Agents in Adults With CKD: A Network Meta-analysis of Randomized Trials. Am J Kidney Dis 68: 691-702. doi: 10.1053/j.ajkd.2016.05.015
|
| [46] |
Raggi P, Bellasi A, Bushinsky D, et al. (2020) Slowing Progression of Cardiovascular Calcification With SNF472 in Patients on Hemodialysis: Results of a Randomized Phase 2b Study. Circulation 141: 728-739. doi: 10.1161/CIRCULATIONAHA.119.044195
|
| [47] |
Raggi P, Chertow GM, Torres PU, et al. (2011) The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 26: 1327-1339. doi: 10.1093/ndt/gfq725
|
| [48] |
Sadek T, Mazouz H, Bahloul H, et al. (2003) Sevelamer hydrochloride with or without alphacalcidol or higher dialysate calcium vs calcium carbonate in dialysis patients: an open-label, randomized study. Nephrol Dial Transplant 18: 582-588. doi: 10.1093/ndt/18.3.582
|
| [49] |
Sawabe M, Arai T, Kasahara I, et al. (2006) Sustained progression and loss of the gender-related difference in atherosclerosis in the very old: a pathological study of 1074 consecutive autopsy cases. Atherosclerosis 186: 374-379. doi: 10.1016/j.atherosclerosis.2005.07.023
|
| [50] |
Dalager S, Paaske WP, Kristensen IB, et al. (2007) Artery-related differences in atherosclerosis expression: implications for atherogenesis and dynamics in intima-media thickness. Stroke 38: 2698-2705. doi: 10.1161/STROKEAHA.107.486480
|
| [51] |
Yahagi K, Kolodgie FD, Otsuka F, et al. (2016) Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol 13: 79-98. doi: 10.1038/nrcardio.2015.164
|
| [52] |
Herisson F, Heymann MF, Chetiveaux M, et al. (2011) Carotid and femoral atherosclerotic plaques show different morphology. Atherosclerosis 216: 348-354. doi: 10.1016/j.atherosclerosis.2011.02.004
|
| [53] |
Derksen WJM, de Vries J-PPM, Vink A, et al. (2010) Histologic atherosclerotic plaque characteristics are associated with restenosis rates after endarterectomy of the common and superficial femoral arteries. J Vasc Surg 52: 592-599. doi: 10.1016/j.jvs.2010.03.063
|
| [54] | Soor GS, Vukin I, Leong SW, et al. (2008) Peripheral vascular disease: who gets it and why? A histomorphological analysis of 261 arterial segments from 58 cases. Pathology (Phila) 40: 385-391. |
| [55] |
Lachman AS, Spray TL, Kerwin DM, et al. (1977) Medial calcinosis of Monckeberg. A review of the problem and a description of a patient with involvement of peripheral, visceral and coronary arteries. Am J Med 63: 615-622. doi: 10.1016/0002-9343(77)90207-8
|
| [56] |
Amos RS, Wright V (1980) Monckeberg's arteriosclerosis and metabolic bone disease. Lancet 2: 248-249. doi: 10.1016/S0140-6736(80)90133-6
|
| [57] | Kolodgie F, Nakazawa G, Santorgi G, et al. (2007) Differences and commons in pathology and reaction on stents between cardiac and peripheral arteries. European Symposium of Vascular Biomaterials 2007 New Technologies in Vascular Biomaterials Strasbourg: EUROPROT 49-70. |
| [58] |
Torii S, Mustapha AJ, Narula J, et al. (2019) Histopathologic Characterization of Peripheral Arteries in Subjects with Abundant Risk Factors. JACC Cardiovasc Imaging 12: 1501-1513. doi: 10.1016/j.jcmg.2018.08.039
|
| [59] |
Mauriello A, Sangiorgi GM, Virmani R, et al. (2010) A pathobiologic link between risk factors profile and morphological markers of carotid instability. Atherosclerosis 208: 572-580. doi: 10.1016/j.atherosclerosis.2009.07.048
|
| [60] | Diehm N, Silvestro A, Baumgartner I, et al. (2009) Chronic critical limb ischemia: European experiences. J Cardiovasc Surg (Torino) 50: 647-653. |
| [61] |
Narula N, Dannenberg AJ, Olin JW, et al. (2018) Pathology of Peripheral Artery Disease in Patients With Critical Limb Ischemia. J Am Coll Cardiol 72: 2152-2163. doi: 10.1016/j.jacc.2018.08.002
|
| [62] |
Kamenskiy A, Poulson W, Sim S, et al. (2018) Prevalence of Calcification in Human Femoropopliteal Arteries and its Association with Demographics, Risk Factors, and Arterial Stiffness. Arterioscler Thromb Vasc Biol 38: e48-e57. doi: 10.1161/ATVBAHA.117.310490
|
| [63] |
Deas DS, Marshall AP, Bian A, et al. (2015) Association of cardiovascular and biochemical risk factors with tibial artery calcification. Vasc Med 20: 326-331. doi: 10.1177/1358863X15581448
|
| [64] |
Shao JS, Cheng SL, Sadhu J, et al. (2010) Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension 55: 579-592. doi: 10.1161/HYPERTENSIONAHA.109.134205
|
| [65] |
Moe SM, Chen NX (2004) Pathophysiology of vascular calcification in chronic kidney disease. Circ Res 95: 560-567. doi: 10.1161/01.RES.0000141775.67189.98
|
| [66] |
Kroger K, Stang A, Kondratieva J, et al. (2006) Prevalence of peripheral arterial disease - results of the Heinz Nixdorf recall study. Eur J Epidemiol 21: 279-285. doi: 10.1007/s10654-006-0015-9
|
| [67] |
Vasuri F, Fittipaldi S, Pacilli A, et al. (2016) The incidence and morphology of Monckeberg's medial calcification in banked vascular segments from a monocentric donor population. Cell Tissue Bank 17: 219-223. doi: 10.1007/s10561-016-9543-z
|
| [68] |
Creager MA, Belkin M, Bluth EI, et al. (2012) 2012 ACCF/AHA/ACR/SCAI/SIR/STS/SVM/SVN/SVS key data elements and definitions for peripheral atherosclerotic vascular disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Clinical Data Standards for Peripheral Atherosclerotic Vascular Disease). Circulation 125: 395-467. doi: 10.1161/CIR.0b013e31823299a1
|
| [69] |
Sahasakul Y, Edwards WD, Naessens JM, et al. (1988) Age-related changes in aortic and mitral valve thickness: implications for two-dimensional echocardiography based on an autopsy study of 200 normal human hearts. Am J Cardiol 62: 424-430. doi: 10.1016/0002-9149(88)90971-X
|
| [70] |
Nkomo VT, Gardin JM, Skelton TN, et al. (2006) Burden of valvular heart diseases: a population-based study. Lancet 368: 1005-1011. doi: 10.1016/S0140-6736(06)69208-8
|
| [71] |
Sakamoto A, Guo L, Virmani R, et al. (2019) Is there a role for activated platelets in progression of aortic valve calcification? Eur Heart J 40: 1374-1377. doi: 10.1093/eurheartj/ehy775
|
| [72] | Butany J, Collins MJ, Demellawy DEI, et al. (2005) Morphological and clinical findings in 247 surgically excised native aortic valves. Can J Cardiol 21: 747-755. |
| [73] |
Roberts WC, Ko JM (2004) Weights of individual cusps in operatively-excised stenotic three-cuspid aortic valves. Am J Cardiol 94: 681-684. doi: 10.1016/j.amjcard.2004.05.045
|
| [74] |
Owens DS, Katz R, Takasu J, et al. (2010) Incidence and progression of aortic valve calcium in the Multi-ethnic Study of Atherosclerosis (MESA). Am J Cardiol 105: 701-708. doi: 10.1016/j.amjcard.2009.10.071
|
| [75] |
Mohler ER, Gannon F, Reynolds C, et al. (2001) Bone formation and inflammation in cardiac valves. Circulation 103: 1522-1528. doi: 10.1161/01.CIR.103.11.1522
|
| [76] |
O'Brien KD, Reichenbach DD, Marcovina SM, et al. (1996) Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol 16: 523-532. doi: 10.1161/01.ATV.16.4.523
|
| [77] |
Milin AC, Vorobiof G, Aksoy O, et al. (2014) Insights into aortic sclerosis and its relationship with coronary artery disease. J Am Heart Assoc 3: 001111. doi: 10.1161/JAHA.114.001111
|
| [78] |
Chiu JJ, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327-387. doi: 10.1152/physrev.00047.2009
|
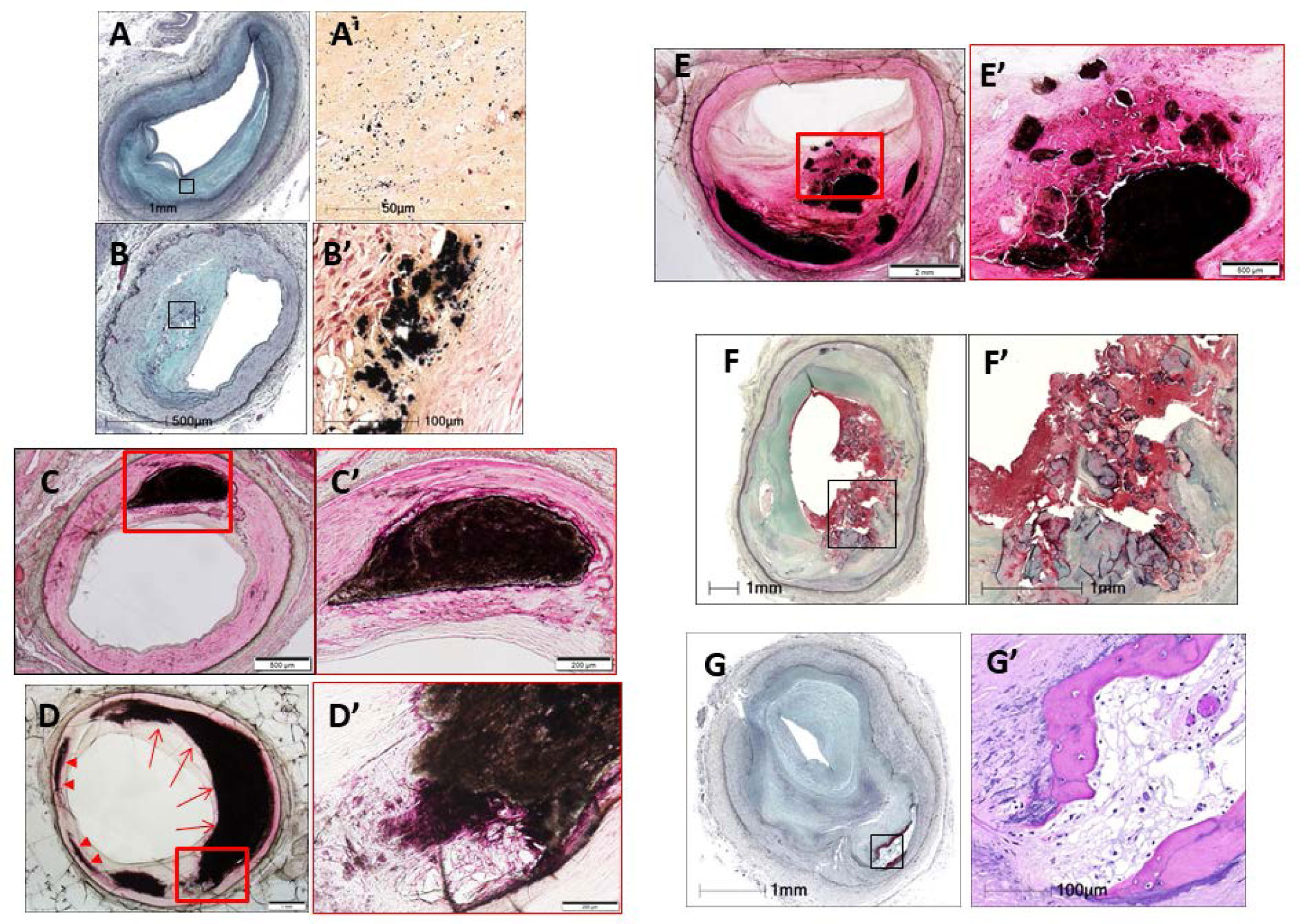
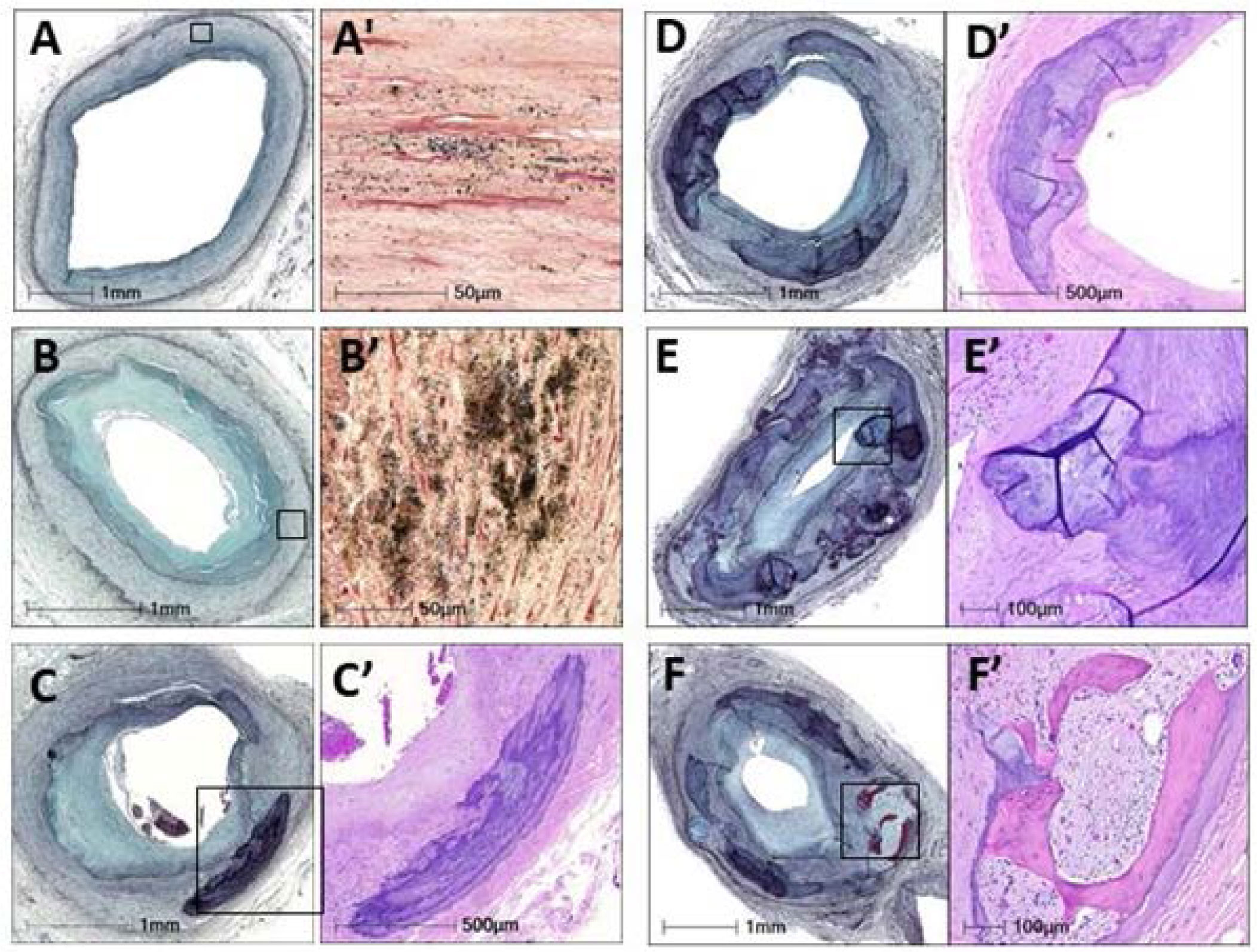
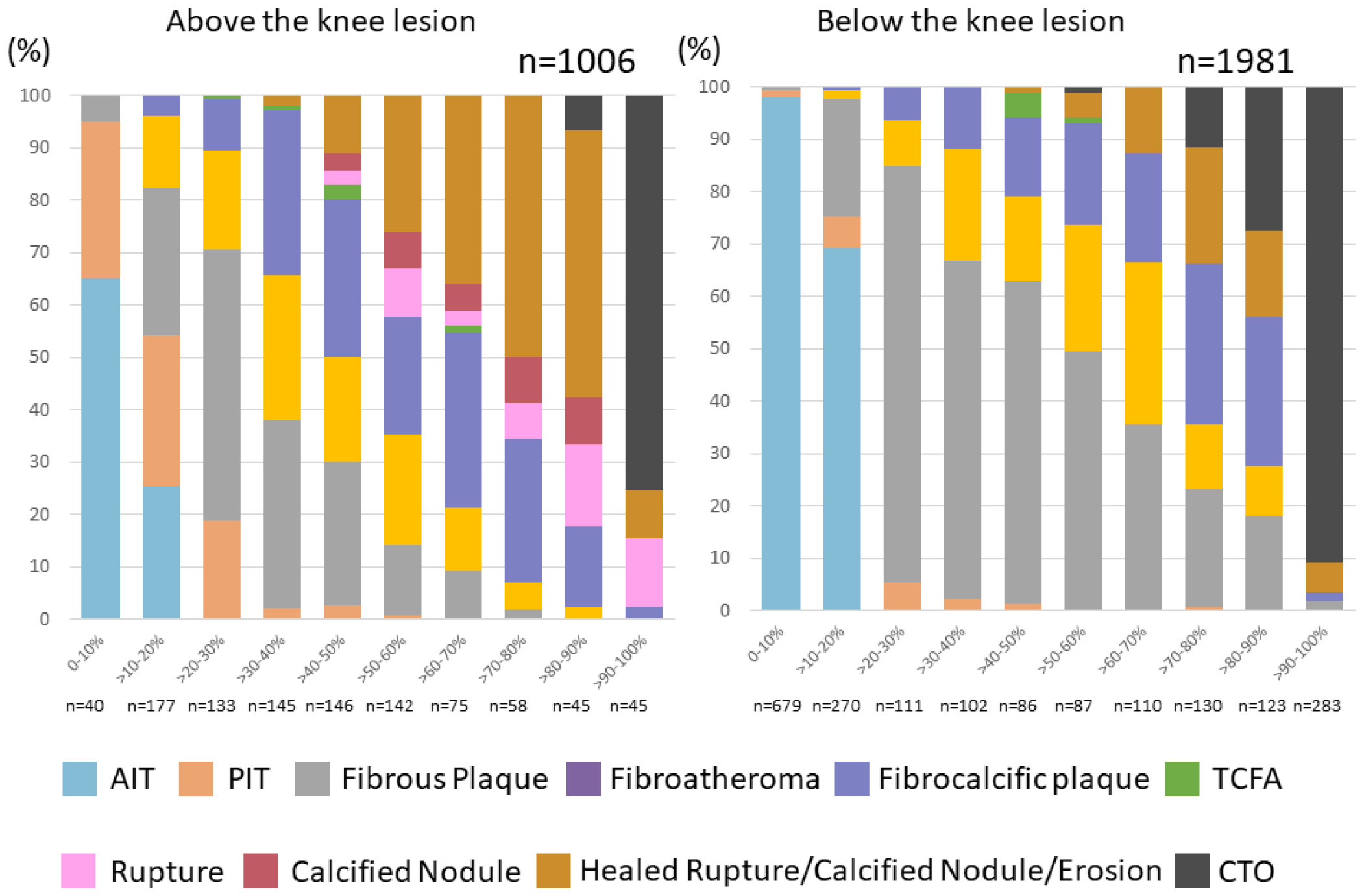
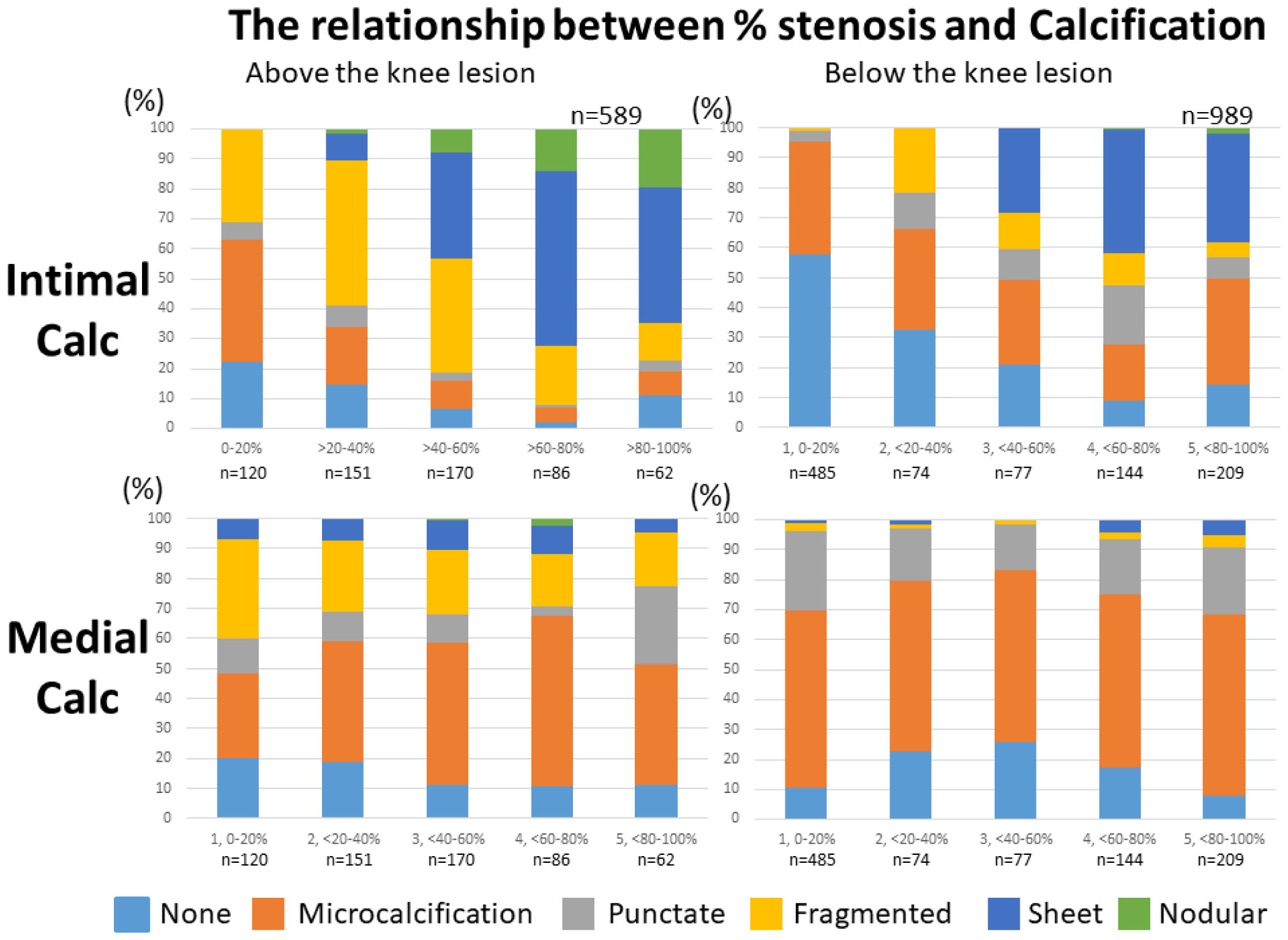
Figures(11)

Yu Sato, Hiroyuki Jinnouchi, Atsushi Sakamoto, Anne Cornelissen, Masayuki Mori, Rika Kawakami, Kenji Kawai, Renu Virmani, Aloke V. Finn. Calcification in human vessels and valves: from pathological point of view[J]. AIMS Molecular Science, 2020, 7(3): 183-210. doi: 10.3934/molsci.2020009







 DownLoad:
DownLoad: