Citation: Manal Alkan, Fadel Sayes, Abdulraouf Ramadan, Francois Machavoine, Michel Dy, Elke Schneider, Nathalie Thieblemont. Basophil activation through TLR2 and TLR4 signaling pathways[J]. AIMS Allergy and Immunology, 2018, 2(3): 126-140. doi: 10.3934/Allergy.2018.3.126

| [1] | Centers for Disease C, Prevention (2011) Vital signs: asthma prevalence, disease characteristics, and self-management education: United States, 2001–2009. MMWR Morb Mortal Wkly Rep 60: 547–552. |
| [2] | Karasuyama H, Miyake K, Yoshikawa S, et al. (2017) Multifaceted roles of basophils in health and disease. J Allergy Clin Immun 6749: 31894–31898. |
| [3] |
Korosec P, Gibbs BF, Rijavec M, et al. (2018) Important and specific role for basophils in acute allergic reactions. Clin Exp Allergy 48: 502–512. doi: 10.1111/cea.13117

|
| [4] | Hashimoto T, Rosen JD, Sanders KM, et al. (2018) Possible roles of basophils in chronic itch. Exp Dermatol, In press. |
| [5] |
Rosenstein RK, Bezbradica JS, Yu S, et al. (2014) Signaling pathways activated by a protease allergen in basophils. Proc Natl Acad Sci USA 111: E4963–E4971. doi: 10.1073/pnas.1418959111
|
| [6] |
Chirumbolo S, Bjorklund G, Sboarina A, et al. (2018) The role of basophils as innate immune regulatory cells in allergy and immunotherapy. Hum Vacc Immunother 14: 815–831. doi: 10.1080/21645515.2017.1417711
|
| [7] |
Lantz CS, Boesiger J, Song CH, et al. (1998) Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature 392: 90–93. doi: 10.1038/32190
|
| [8] | Le Gros G, Ben-Sasson SZ, Conrad DH, et al. (1990) IL-3 promotes production of IL-4 by splenic non-B, non-T cells in response to Fc receptor cross-linkage. J Immunol 145: 2500–2506. |
| [9] |
Min B, Prout M, Hu LJ, et al. (2004) Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med 200: 507–517. doi: 10.1084/jem.20040590
|
| [10] |
Hida S, Yamasaki S, Sakamoto Y, et al. (2009) Fc receptor gamma-chain, a constitutive component of the IL-3 receptor, is required for IL-3-induced IL-4 production in basophils. Nat Immunol 10: 214–222. doi: 10.1038/ni.1686
|
| [11] |
Ohmori K, Luo Y, Jia Y, et al. (2009) IL-3 induces basophil expansion in vivo by directing granulocyte-monocyte progenitors to differentiate into basophil lineage-restricted progenitors in the bone marrow and by increasing the number of basophil/mast cell progenitors in the spleen. J Immunol 182: 2835–2841. doi: 10.4049/jimmunol.0802870
|
| [12] |
Herbst T, Esser J, Prati M, et al. (2012) Antibodies and IL-3 support helminth-induced basophil expansion. Proc Natl Acad Sci USA 109: 14954–14959. doi: 10.1073/pnas.1117584109
|
| [13] |
Siracusa MC, Saenz SA, Hill DA, et al. (2011) TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 477: 229–233. doi: 10.1038/nature10329
|
| [14] |
Schneider E, Petit-Bertron AF, Bricard R, et al. (2009) IL-33 activates unprimed murine basophils directly in vitro and induces their in vivo expansion indirectly by promoting hematopoietic growth factor production. J Immunol 183: 3591–3597. doi: 10.4049/jimmunol.0900328
|
| [15] |
Varricchi G, Raap U, Rivellese F, et al. (2018) Human mast cells and basophils-How are they similar how are they different? Immunol Rev 282: 8–34. doi: 10.1111/imr.12627
|
| [16] |
Ohnmacht C, Voehringer D (2010) Basophils protect against reinfection with hookworms independently of mast cells and memory Th2 cells. J Immunol 184: 344–350. doi: 10.4049/jimmunol.0901841
|
| [17] |
Kampfer SS, Odermatt A, Dahinden CA, et al. (2017) Late IL-3-induced phenotypic and functional alterations in human basophils require continuous IL-3 receptor signaling. J Leukocyte Biol 101: 227–238. doi: 10.1189/jlb.2A0715-292RR
|
| [18] |
Reese TA, Liang HE, Tager AM, et al. (2007) Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature 447: 92–96. doi: 10.1038/nature05746
|
| [19] | Schneider E, Thieblemont N, De Moraes ML, et al. (2010) Basophils: new players in the cytokine network. Eur Cytokine Netw 21: 142–153. |
| [20] |
Brown GD, Herre J, Williams DL, et al. (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197: 1119–1124. doi: 10.1084/jem.20021890
|
| [21] |
Ramadan A, Pham VL, Machavoine F, et al. (2013) Activation of basophils by the double-stranded RNA poly(A:U) exacerbates allergic inflammation. Allergy 68: 732–738. doi: 10.1111/all.12151
|
| [22] |
Hill DA, Siracusa MC, Abt MC, et al. (2012) Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med 18: 538–546. doi: 10.1038/nm.2657
|
| [23] |
Hammad H, Chieppa M, Perros F, et al. (2009) House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med 15: 410–416. doi: 10.1038/nm.1946
|
| [24] |
Iwasaki N, Matsushita K, Fukuoka A, et al. (2017) Allergen endotoxins induce T-cell-dependent and non-IgE-mediated nasal hypersensitivity in mice. J Allergy Clin Immun 139: 258–268.e210. doi: 10.1016/j.jaci.2016.03.023
|
| [25] |
Schroeder JT, Chichester KL, Bieneman AP (2009) Human basophils secrete IL-3: evidence of autocrine priming for phenotypic and functional responses in allergic disease. J Immunol 182: 2432–2438. doi: 10.4049/jimmunol.0801782
|
| [26] |
Gentinetta T, Pecaric-Petkovic T, Wan D, et al. (2011) Individual IL-3 priming is crucial for consistent in vitro activation of donor basophils in patients with chronic urticaria. J Allergy Clin Immun 128: 1227–1234.e1225. doi: 10.1016/j.jaci.2011.07.021
|
| [27] |
Bieneman AP, Chichester KL, Chen YH, et al. (2005) Toll-like receptor 2 ligands activate human basophils for both IgE-dependent and IgE-independent secretion. J Allergy Clin Immun 115: 295–301. doi: 10.1016/j.jaci.2004.10.018
|
| [28] | Smith TF, Aelvoet M, Morrison DC (1985) The effect of bacterial lipopolysaccharide (LPS) on histamine release from human basophils. I. Enhancement of immunologic release by LPS. Clin Immunol Immunopathol 34: 355–365. |
| [29] | Norn S, Baek L, Jensen C, et al. (1986) Influence of bacterial endotoxins on basophil histamine release. Potentiation of antigen- and bacteria-induced histamine release. Allergy 41: 125–130. |
| [30] |
Schwartz C, Oeser K, Prazeres da CC, et al. (2014) T cell-derived IL-4/IL-13 protects mice against fatal Schistosoma mansoni infection independently of basophils. J Immunol 193: 3590–3599. doi: 10.4049/jimmunol.1401155
|
| [31] |
Magalhaes K, Almeida PE, Atella G, et al. (2010) Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through Toll-like receptor-2-dependent mechanisms. J Infect Dis 202: 1369–1379. doi: 10.1086/656477
|
| [32] |
Watanabe T, Yamashita K, Sakurai T, et al. (2013) Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol 48: 247–253. doi: 10.1007/s00535-012-0626-8
|
| [33] |
Wenceslau CF, McCarthy CG, Szasz T, et al. (2015) Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol-Heart C 308: H768–H777. doi: 10.1152/ajpheart.00779.2014
|
| [34] |
Yamanishi Y, Miyake K, Iki M, et al. (2017) Recent advances in understanding basophil-mediated Th2 immune responses. Immunol Rev 278: 237–245. doi: 10.1111/imr.12548
|
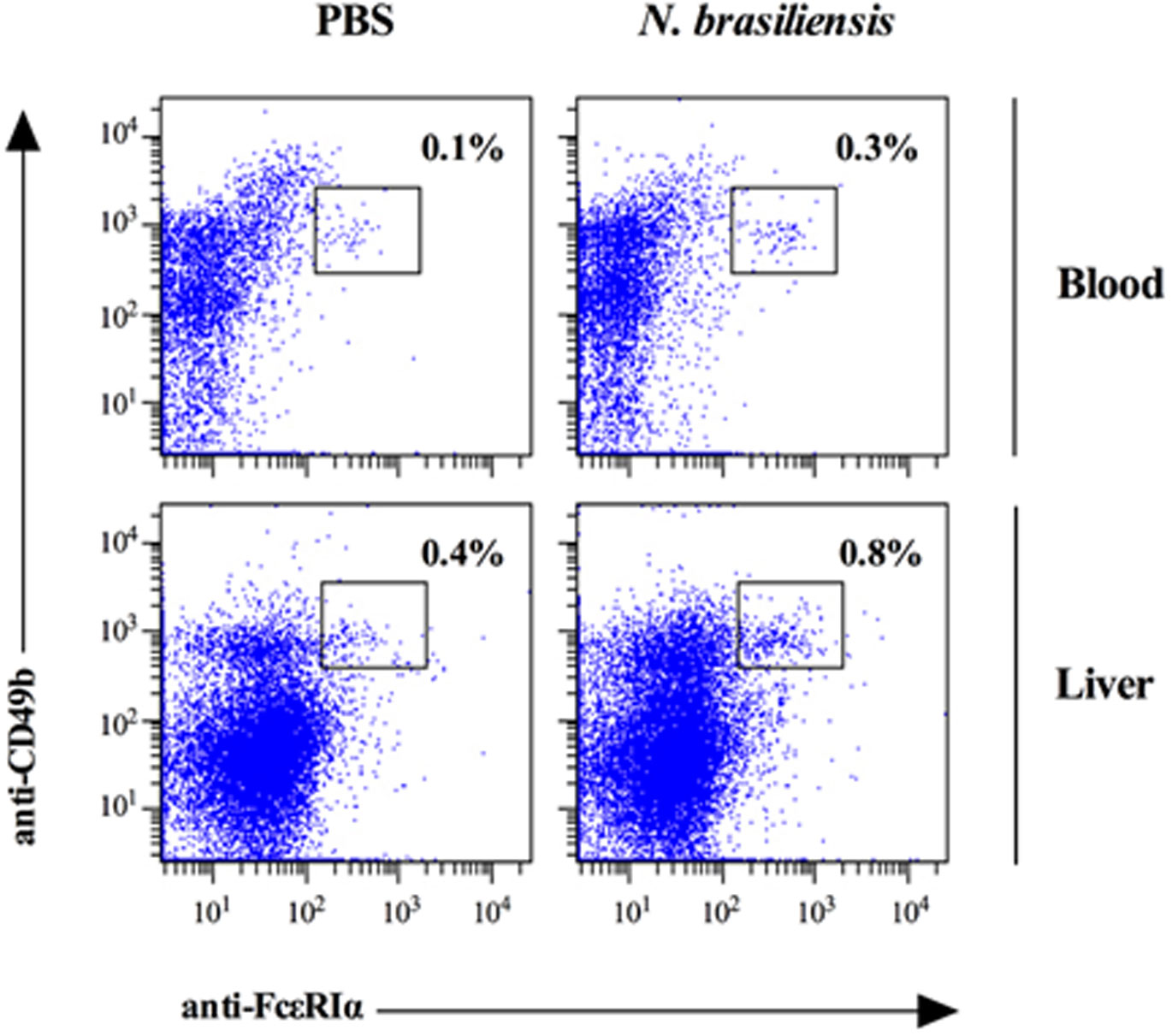
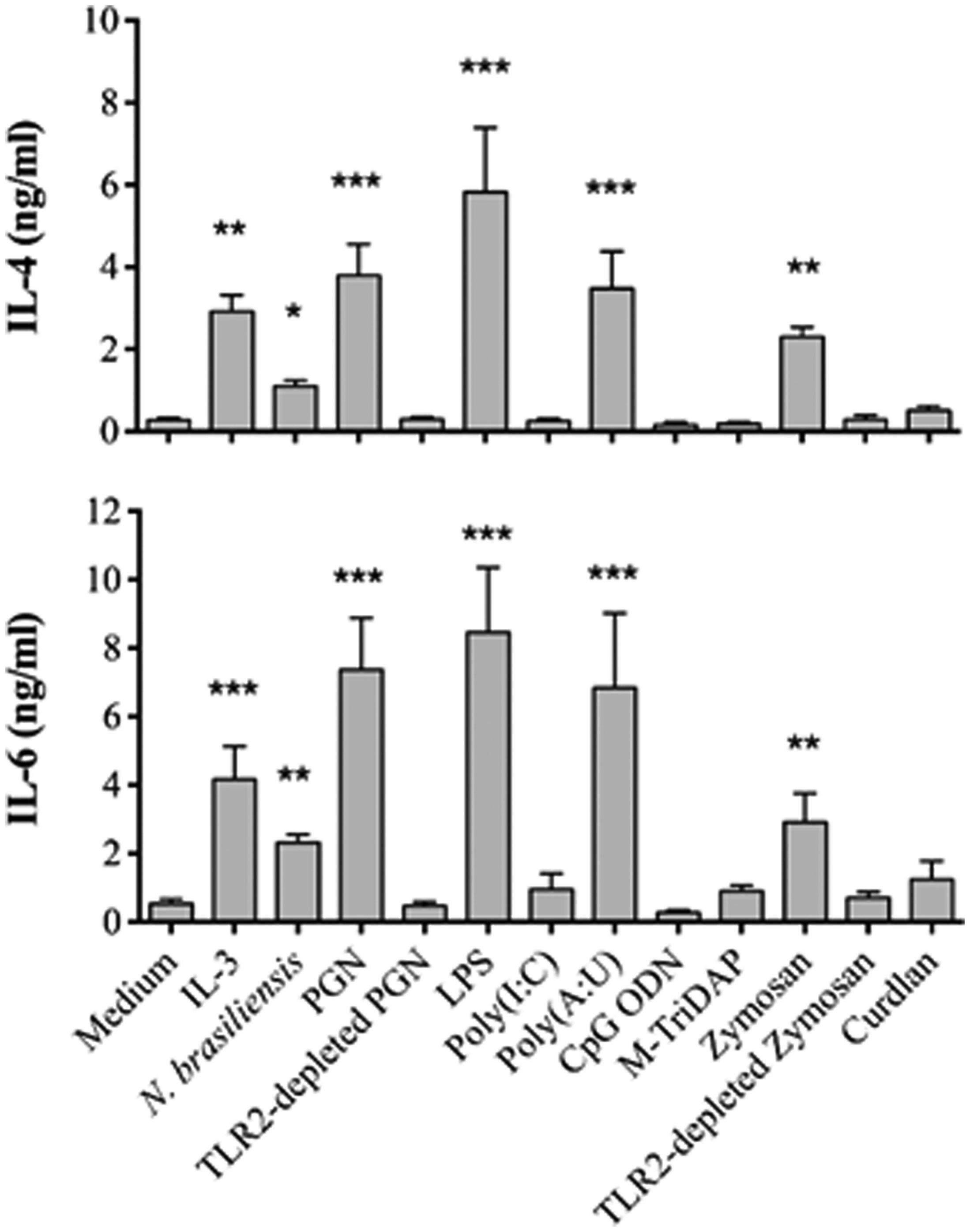
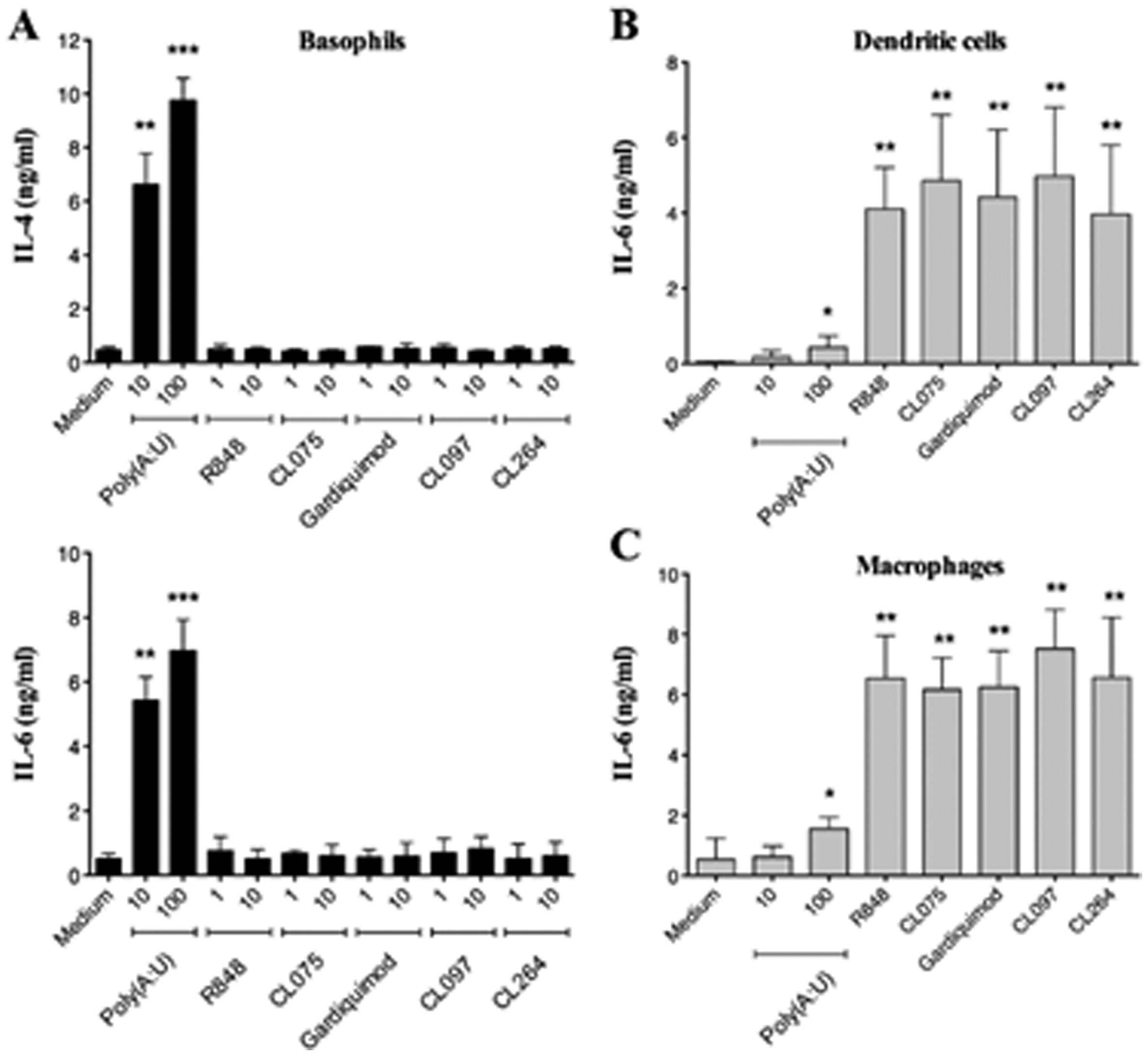
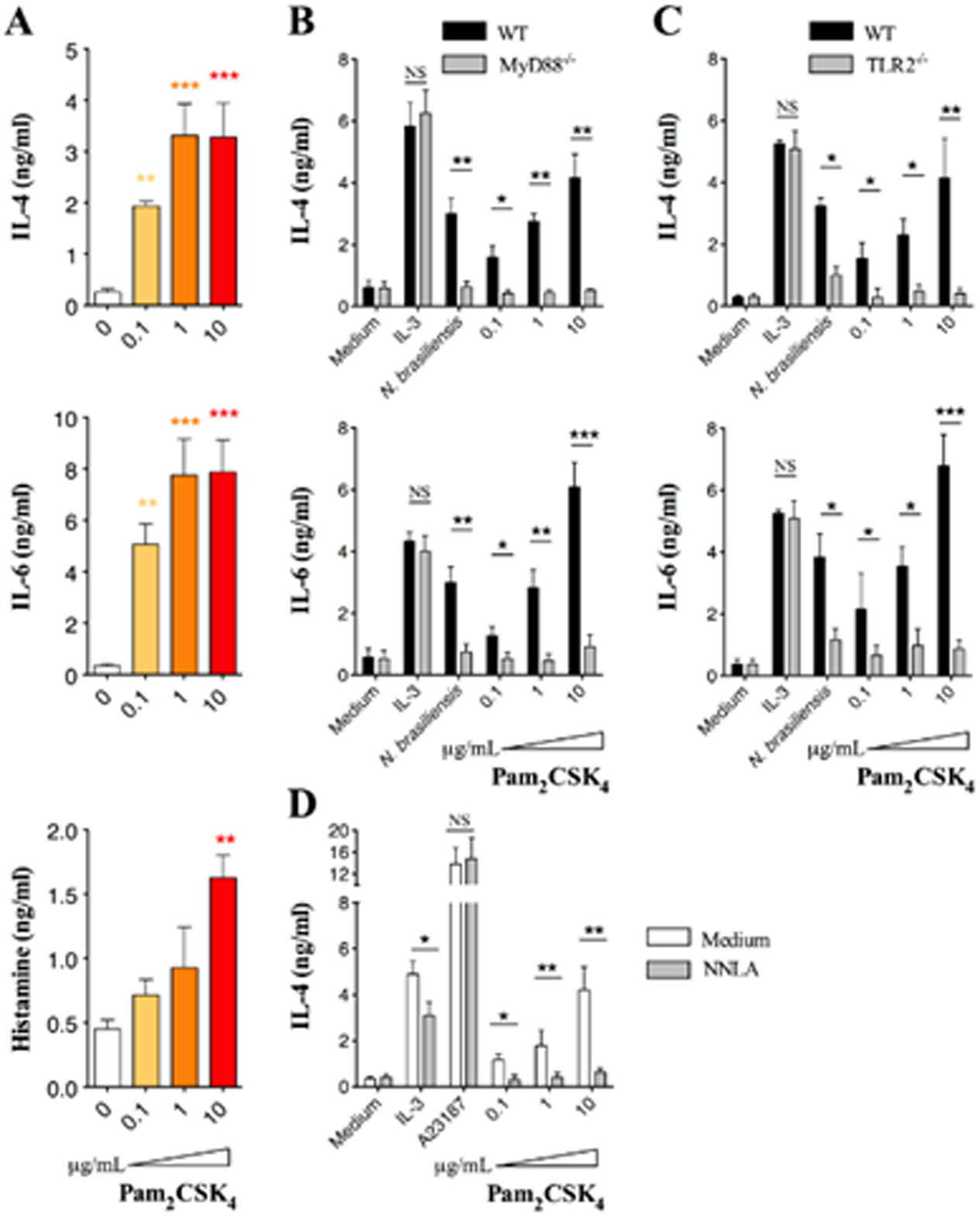
Figures(7)

Manal Alkan, Fadel Sayes, Abdulraouf Ramadan, Francois Machavoine, Michel Dy, Elke Schneider, Nathalie Thieblemont. Basophil activation through TLR2 and TLR4 signaling pathways[J]. AIMS Allergy and Immunology, 2018, 2(3): 126-140. doi: 10.3934/Allergy.2018.3.126







 DownLoad:
DownLoad: