Citation: Charles A. Downs, Abdel A. Alli, Nicholle M. Johnson, My N. Helms. Cigarette smoke extract is a Nox agonist and regulates ENaC in alveolar type 2 cells[J]. AIMS Molecular Science, 2016, 3(3): 439-453. doi: 10.3934/molsci.2016.3.439

| [1] |
Eaton DC, Helms MN, Koval M, et al. (2009) The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 71: 403-423. doi: 10.1146/annurev.physiol.010908.163250

|
| [2] |
Hummler E, Barker P, Gatzy J, et al. (1996) Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet 12: 325-328. doi: 10.1038/ng0396-325
|
| [3] |
Hummler E, Barker P, Talbot C, et al. (1997) A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc Natl Acad Sci U S A 94: 11710-11715. doi: 10.1073/pnas.94.21.11710
|
| [4] |
Mall M, Grubb BR, Harkema JR, et al. (2004) Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487-493. doi: 10.1038/nm1028
|
| [5] |
Mall MA, Harkema JR, Trojanek JB, et al. (2008) Development of chronic bronchitis and emphysema in beta-epithelial Na+ channel-overexpressing mice. Am J Respir Crit Care Med 177: 730-742. doi: 10.1164/rccm.200708-1233OC
|
| [6] |
Mall MA (2009) Role of the amiloride-sensitive epithelial Na+ channel in the pathogenesis and as a therapeutic target for cystic fibrosis lung disease. Exp Physiol 94: 171-174. doi: 10.1113/expphysiol.2008.042994
|
| [7] |
Downs CA, Kreiner LH, Johnson NM, et al. (2015) Receptor for advanced glycation end-products regulates lung fluid balance via protein kinase C-gp91(phox) signaling to epithelial sodium channels. Am J Respir Cell Mol Biol 52: 75-87. doi: 10.1165/rcmb.2014-0002OC
|
| [8] |
Goodson P, Kumar A, Jain L, et al. (2012) Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O⁻₂ signaling. Am J Physiol Lung Cell Mol Physiol 302: L410-419. doi: 10.1152/ajplung.00260.2011
|
| [9] |
Aguirre J, Lambeth JD (2010) Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic Biol Med 49: 1342-1353. doi: 10.1016/j.freeradbiomed.2010.07.027
|
| [10] |
Babior BM (2004) NADPH oxidase. Curr Opin Immunol 16: 42-47. doi: 10.1016/j.coi.2003.12.001
|
| [11] |
Geiszt M (2006) NADPH oxidases: new kids on the block. Cardiovasc Res 71: 289-299. doi: 10.1016/j.cardiores.2006.05.004
|
| [12] |
Takemura Y, Goodson P, Bao HF, et al. (2010) Rac1-mediated NADPH oxidase release of O2- regulates epithelial sodium channel activity in the alveolar epithelium. Am J Physiol Lung Cell Mol Physiol 298: L509-520. doi: 10.1152/ajplung.00230.2009
|
| [13] |
Trac D, Liu B, Pao AC, et al. (2013) Fulvene-5 inhibition of Nadph oxidases attenuates activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 305: F995-F1005. doi: 10.1152/ajprenal.00098.2013
|
| [14] |
Barnes PJ (2014) Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35: 71-86. doi: 10.1016/j.ccm.2013.10.004
|
| [15] |
Rahman I (2005) Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys 43: 167-188. doi: 10.1385/CBB:43:1:167
|
| [16] |
Downs CA, Kreiner LH, Trac DQ, et al. (2013) Acute effects of cigarette smoke extract on alveolar epithelial sodium channel activity and lung fluid clearance. Am J Respir Cell Mol Biol 49: 251-259. doi: 10.1165/rcmb.2012-0234OC
|
| [17] |
Helms MN, Self J, Bao HF, et al. (2006) Dopamine activates amiloride-sensitive sodium channels in alveolar type I cells in lung slice preparations. Am J Physiol Lung Cell Mol Physiol 291: L610-618. doi: 10.1152/ajplung.00426.2005
|
| [18] |
Downs CA, Montgomery DW, Merkle CJ (2011) Age-related differences in cigarette smoke extract-induced H2O2 production by lung endothelial cells. Microvasc Res 82: 311-317. doi: 10.1016/j.mvr.2011.09.013
|
| [19] |
Downs CA, Kreiner L, Zhao XM, et al. (2015) Oxidized glutathione (GSSG) inhibits epithelial sodium channel activity in primary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 308: L943-952. doi: 10.1152/ajplung.00213.2014
|
| [20] |
Yu L, Helms MN, Yue Q, et al. (2008) Single-channel analysis of functional epithelial sodium channel (ENaC) stability at the apical membrane of A6 distal kidney cells. Am J Physiol Renal Physiol 295: F1519-1527. doi: 10.1152/ajprenal.00605.2007
|
| [21] |
Helms MN, Jain L, Self JL, et al. (2008) Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J Biol Chem 283: 22875-22883. doi: 10.1074/jbc.M801363200
|
| [22] |
Church DF, Pryor WA (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64: 111-126. doi: 10.1289/ehp.8564111
|
| [23] |
Pryor WA, Prier DG, Church DF (1983) Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect 47: 345-355. doi: 10.1289/ehp.8347345
|
| [24] |
Pryor WA, Hales BJ, Premovic PI, et al. (1983) The radicals in cigarette tar: their nature and suggested physiological implications. Science 220: 425-427. doi: 10.1126/science.6301009
|
| [25] |
Pryor WA, Church DF, Evans MD, et al. (1990) A comparison of the free radical chemistry of tobacco-burning cigarettes and cigarettes that only heat tobacco. Free Radic Biol Med 8: 275-279. doi: 10.1016/0891-5849(90)90075-T
|
| [26] |
Buttigieg J, Pan J, Yeger H, et al. (2012) NOX2 (gp91phox) is a predominant O2 sensor in a human airway chemoreceptor cell line: biochemical, molecular, and electrophysiological evidence. Am J Physiol Lung Cell Mol Physiol 303: L598-607. doi: 10.1152/ajplung.00170.2012
|
| [27] |
Nisimoto Y, Diebold BA, Cosentino-Gomes D, et al. (2014) Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111-5120. doi: 10.1021/bi500331y
|
| [28] |
Chen M, Xiao D, Hu XQ, et al. (2015) Hypoxia Represses ER-alpha Expression and Inhibits Estrogen-Induced Regulation of Ca2+-Activated K+ Channel Activity and Myogenic Tone in Ovine Uterine Arteries: Causal Role of DNA Methylation. Hypertension 66: 44-51. doi: 10.1161/HYPERTENSIONAHA.115.05299
|
| [29] | Lai N, Lu W, Wang J (2015) Ca(2+) and ion channels in hypoxia-mediated pulmonary hypertension. Int J Clin Exp Pathol 8: 1081-1092. |
| [30] |
Downs CA, Trac DQ, Kreiner LH, et al. (2013) Ethanol alters alveolar fluid balance via Nadph oxidase (NOX) signaling to epithelial sodium channels (ENaC) in the lung. PLoS One 8: e54750. doi: 10.1371/journal.pone.0054750
|
| [31] |
Downs CA, Kumar A, Kreiner LH, et al. (2013) H2O2 regulates lung epithelial sodium channel (ENaC) via ubiquitin-like protein Nedd8. J Biol Chem 288: 8136-8145. doi: 10.1074/jbc.M112.389536
|
| [32] |
Downs CA, Helms MN (2013) Regulation of ion transport by oxidants. Am J Physiol Lung Cell Mol Physiol 305: L595-603. doi: 10.1152/ajplung.00212.2013
|
| [33] |
Peters DM, Vadász I, Wujak L, et al. (2014) TGF-β directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci U S A 111: E374-383. doi: 10.1073/pnas.1306798111
|
| [34] |
von Löhneysen K, Noack D, Jesaitis AJ, et al. (2008) Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem 283: 35273-35282. doi: 10.1074/jbc.M804200200
|
| [35] |
Amara N, Goven D, Prost F, et al. (2010) NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 65: 733-738. doi: 10.1136/thx.2009.113456
|
| [36] |
Boudreau HE, Casterline BW, Burke DJ, et al. (2014) Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Br J Cancer 110: 2569-2582. doi: 10.1038/bjc.2014.165
|
| [37] |
Crestani B, Besnard V, Boczkowski J (2011) Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 43: 1086-1089. doi: 10.1016/j.biocel.2011.04.003
|
| [38] |
Pache JC, Carnesecchi S, Deffert C, et al. (2011) NOX-4 is expressed in thickened pulmonary arteries in idiopathic pulmonary fibrosis. Nat Med 17: 31-32. doi: 10.1038/nm0111-31
|
| [39] |
Yan S, Liu G, Pei C, et al. (2015) Inhibition of NADPH oxidase protects against metastasis of human lung cancer by decreasing microRNA-21. Anticancer Drugs 26: 388-398. doi: 10.1097/CAD.0000000000000198
|
| [40] |
Zinellu A, Fois AG, Sotgia S, et al. (2016) Plasma protein thiols: an early marker of oxidative stress in asthma and chronic obstructive pulmonary disease. Eur J Clin Invest 46: 181-188. doi: 10.1111/eci.12582
|
| [41] |
Cantin AM, Hanrahan JW, Bilodeau G, et al. (2006) Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med 173: 1139-1144. doi: 10.1164/rccm.200508-1330OC
|
| [42] |
Kreindler JL, Jackson AD, Kemp PA, et al. (2005) Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am J Physiol Lung Cell Mol Physiol 288: L894-902. doi: 10.1152/ajplung.00376.2004
|
| [43] | Sloane P A, Shastry S, Wilhelm A, et al. (2012) A Pharmacological Approach to Acquired Cystic Fibrosis Transmembrane Conductance Regulator Dysfunction in Smoking Related Lung Disease. PloS one 7. |
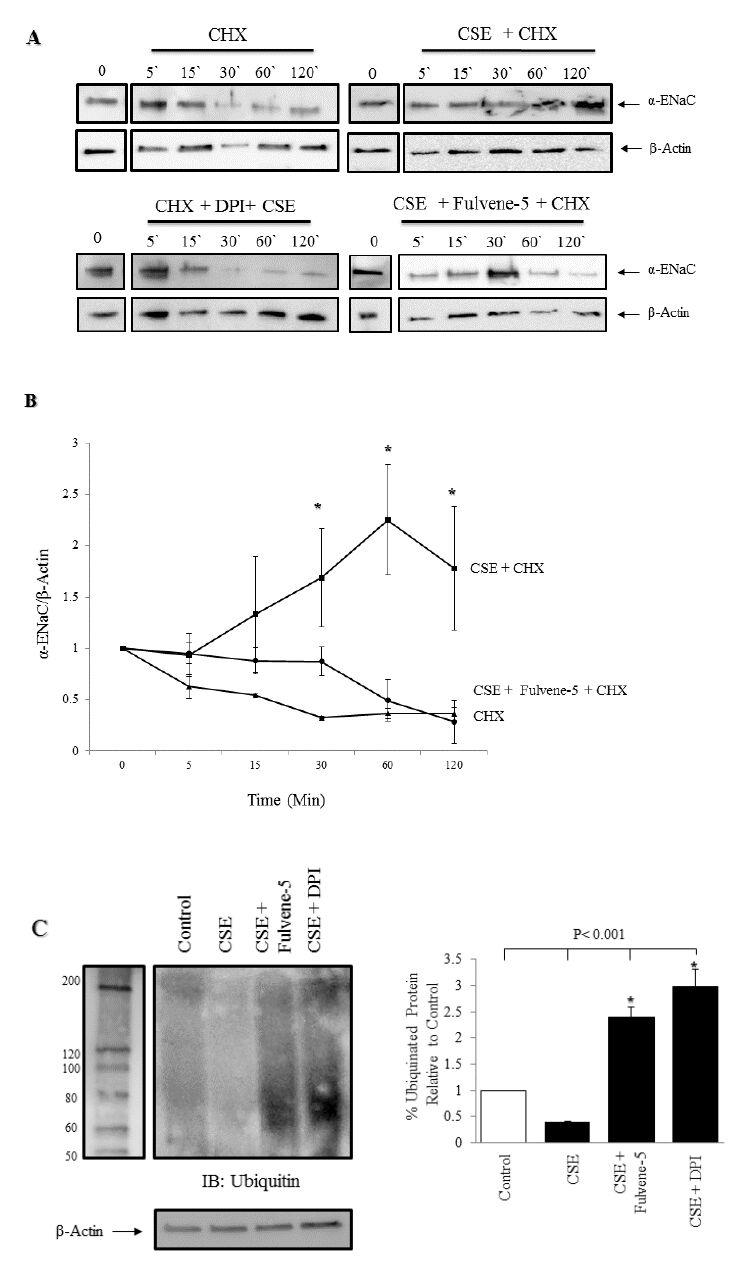
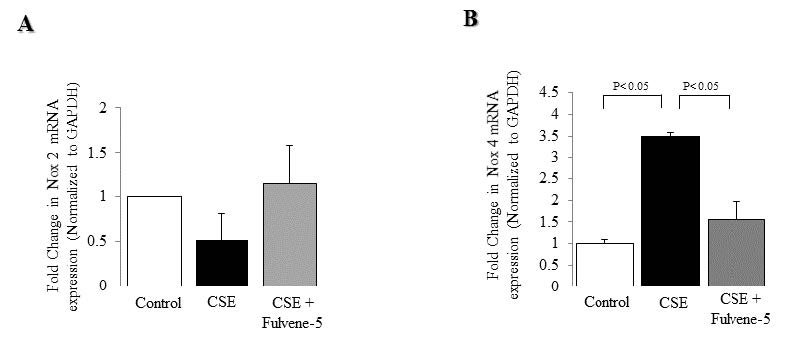
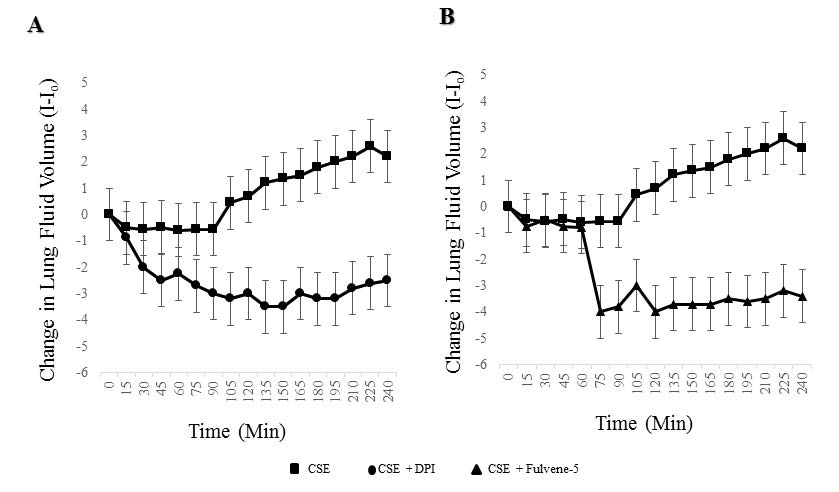
Figures(7)

Charles A. Downs, Abdel A. Alli, Nicholle M. Johnson, My N. Helms. Cigarette smoke extract is a Nox agonist and regulates ENaC in alveolar type 2 cells[J]. AIMS Molecular Science, 2016, 3(3): 439-453. doi: 10.3934/molsci.2016.3.439







 DownLoad:
DownLoad: