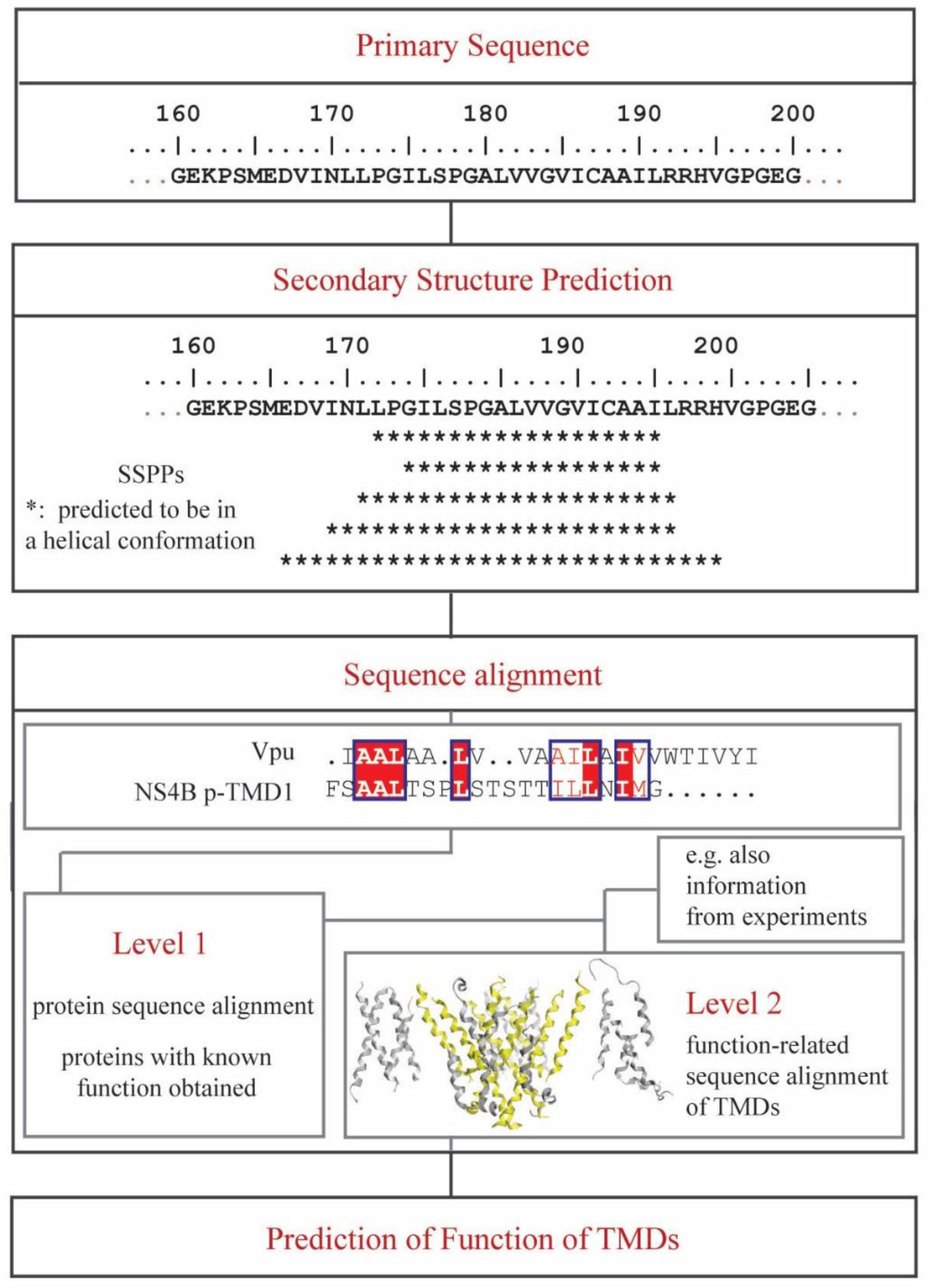
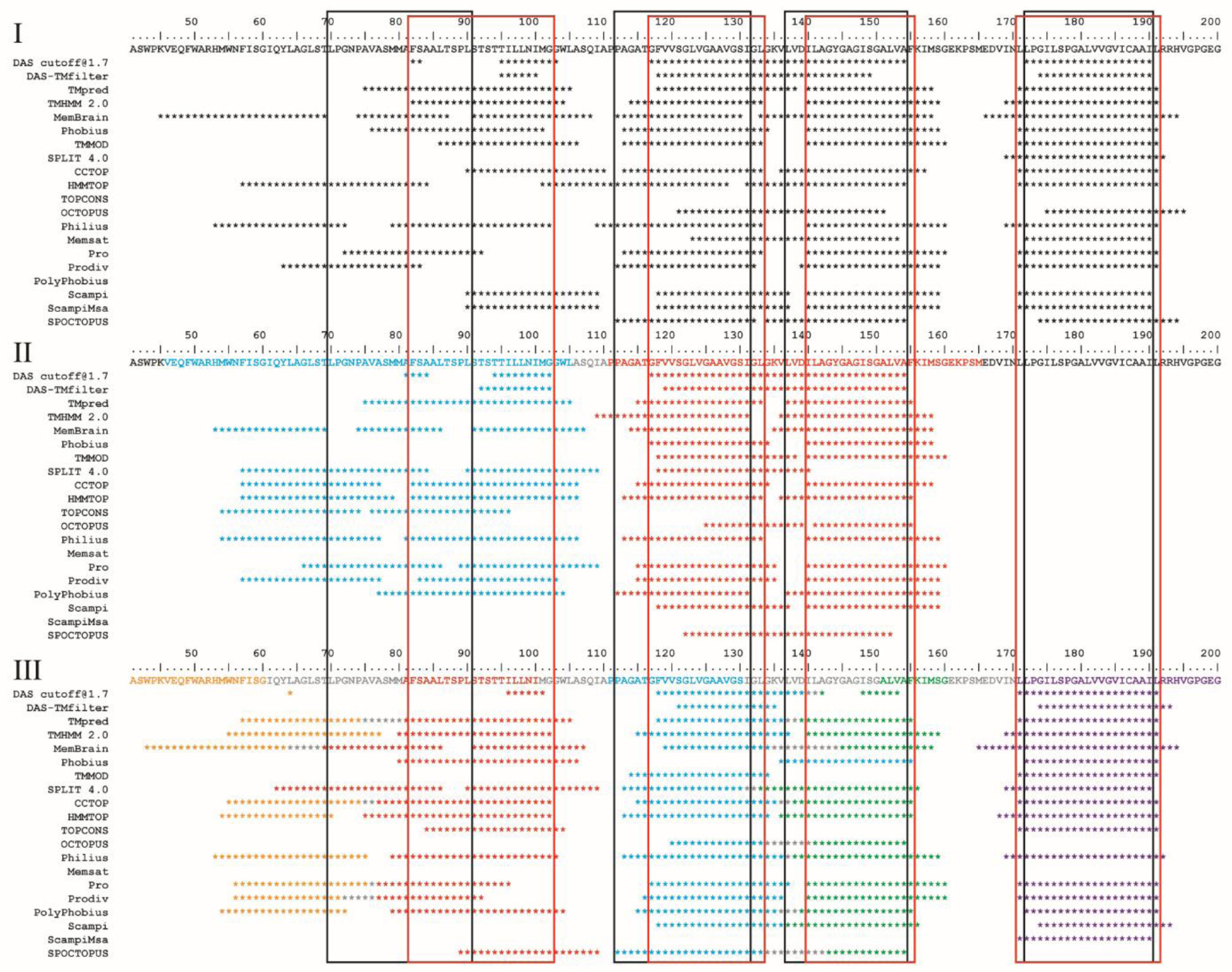
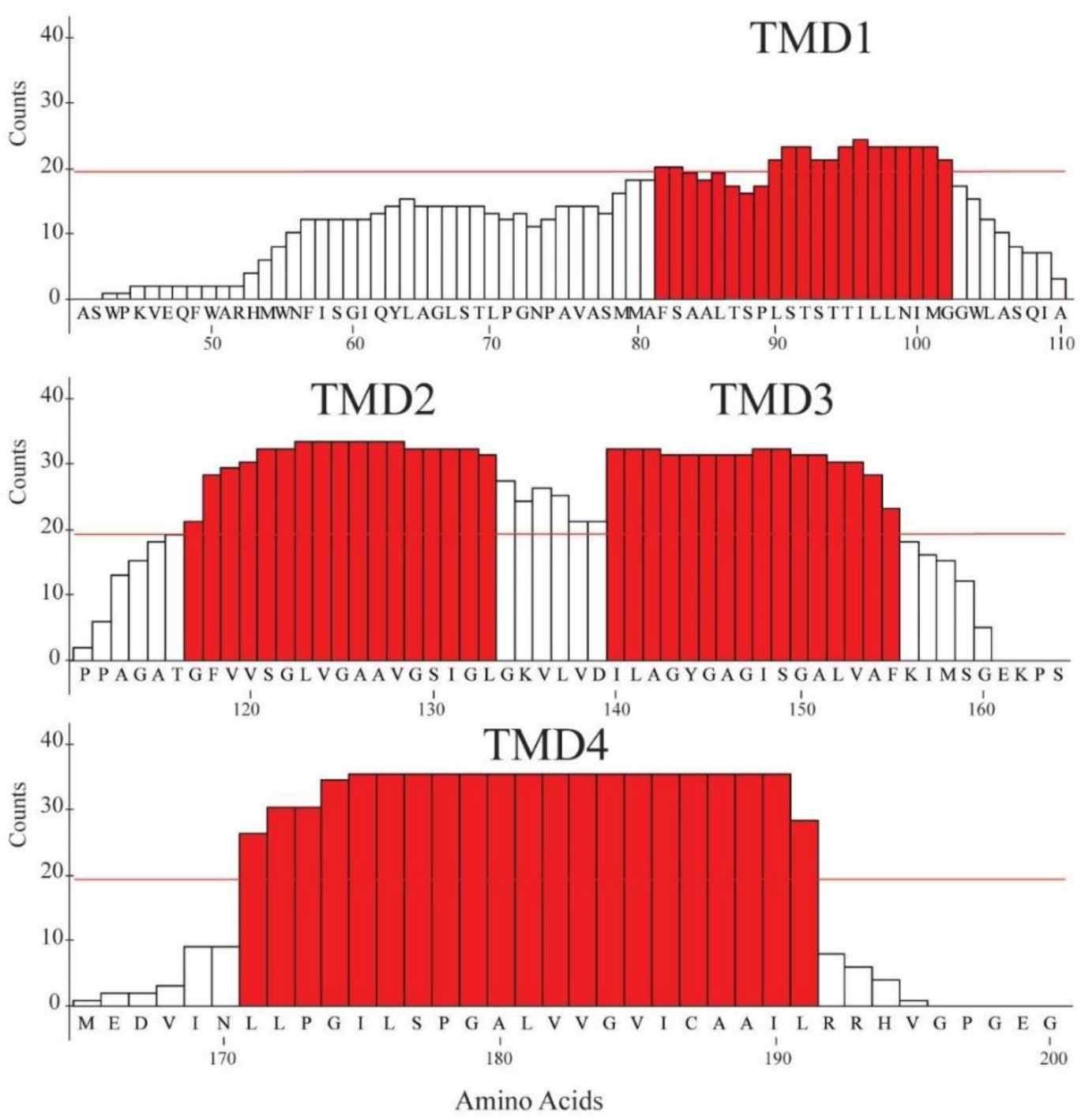
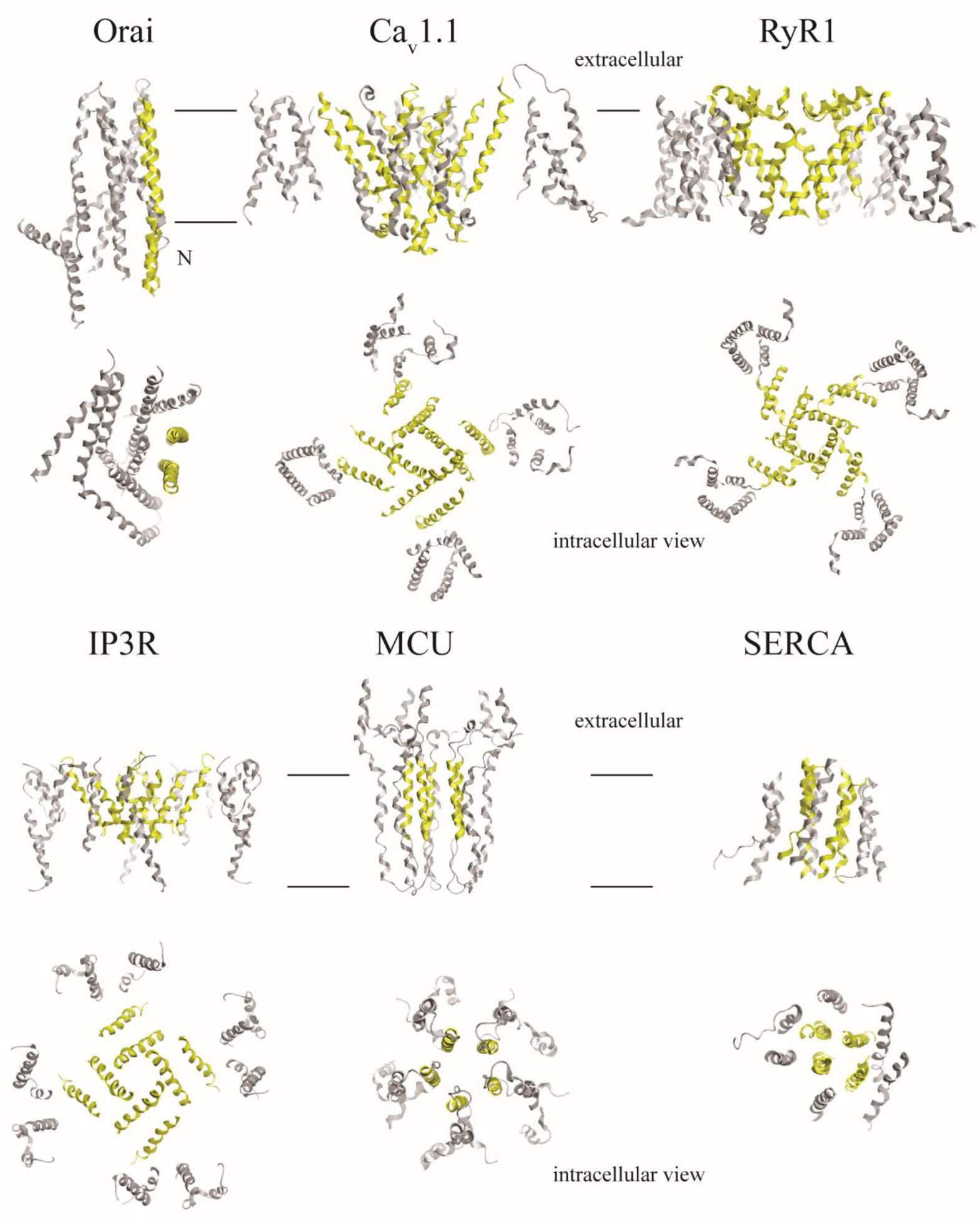
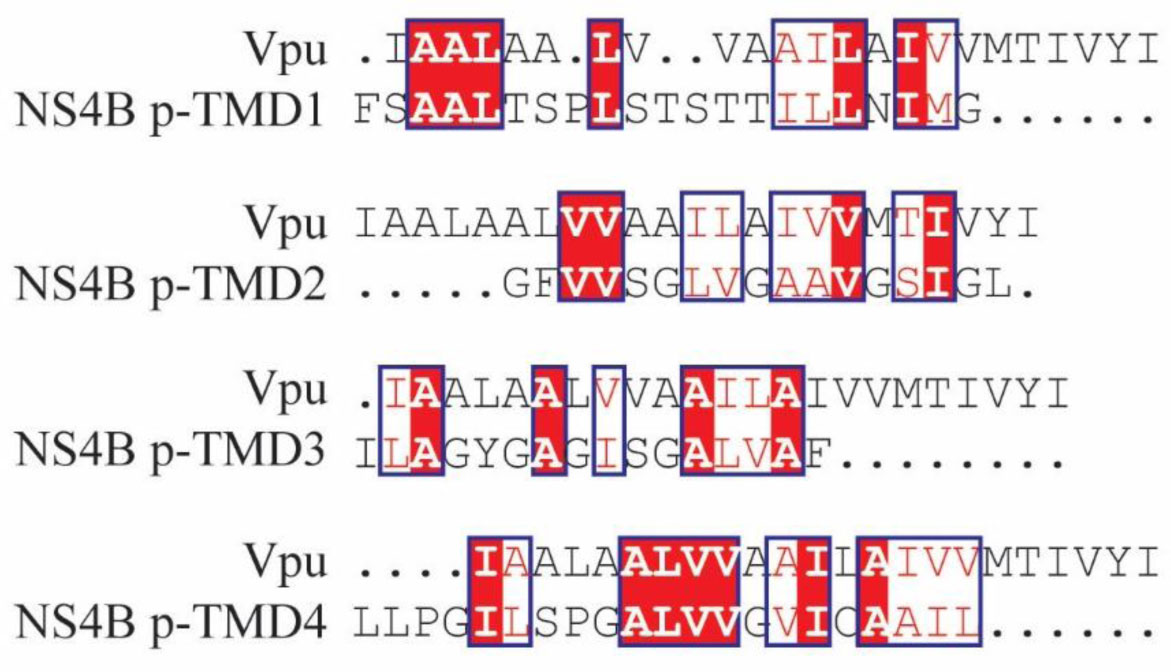
An algorithm is applied to propose a sequence–function correlation of the transmembrane domains (TMDs) of the non-structural protein 4B (NS4B) of hepatitis C virus (HCV). The putative sequence of the TMDs is obtained using 20 available secondary structure prediction programs (SSPPs) with different lengths of the overall amino acid sequence of the protein as input. The results support the notion of four helical TMDs. Whilst the region of the first TMDs leaves room for speculation about an additional TMD, the other three TMDs are consistently predicted. Structural features and the role of each of the TMDs is proposed by applying pairwise sequence alignment using BLAST on the level (i) protein sequence alignment and consequent (ii) function-related alignment. Sequence identity with those TMDs of proteins involved in Ca-homeostasis and generation of replication vesicles, such as Nsp3 of corona viruses, murine coronavirus especially mouse hepatitis virus (MHV), middle east respiratory syndrome coronavirus (MERS), severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2, are suggested. Focusing the search on those proteins in particular and their TMDs playing an active role in their mechanism of function, such as transporters, pumps, viral channel forming protein Vpu of human immunodeficiency virus type 1 (HIV-1) and mediators, suggests TMDs 2 and 4 to have functional roles in NS4B, as well as additionally TMD1 and 3 in case of vesicle formation.
Citation: Ta-Chou Huang, Wolfgang B. Fischer. Sequence–function correlation of the transmembrane domains in NS4B of HCV using a computational approach[J]. AIMS Biophysics, 2021, 8(2): 165-181. doi: 10.3934/biophy.2021013
An algorithm is applied to propose a sequence–function correlation of the transmembrane domains (TMDs) of the non-structural protein 4B (NS4B) of hepatitis C virus (HCV). The putative sequence of the TMDs is obtained using 20 available secondary structure prediction programs (SSPPs) with different lengths of the overall amino acid sequence of the protein as input. The results support the notion of four helical TMDs. Whilst the region of the first TMDs leaves room for speculation about an additional TMD, the other three TMDs are consistently predicted. Structural features and the role of each of the TMDs is proposed by applying pairwise sequence alignment using BLAST on the level (i) protein sequence alignment and consequent (ii) function-related alignment. Sequence identity with those TMDs of proteins involved in Ca-homeostasis and generation of replication vesicles, such as Nsp3 of corona viruses, murine coronavirus especially mouse hepatitis virus (MHV), middle east respiratory syndrome coronavirus (MERS), severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2, are suggested. Focusing the search on those proteins in particular and their TMDs playing an active role in their mechanism of function, such as transporters, pumps, viral channel forming protein Vpu of human immunodeficiency virus type 1 (HIV-1) and mediators, suggests TMDs 2 and 4 to have functional roles in NS4B, as well as additionally TMD1 and 3 in case of vesicle formation.

| [1] | KcDB (2017) Recent advances in sequence-based protein structure prediction. Brief Bioinform 18: 1021-1032. |
| [2] |
Rost B, Sander C (1996) Bridging the protein sequence-structure gap by structure predictions. Annu Rev Biophys Biomol Struct 25: 113-136. doi: 10.1146/annurev.bb.25.060196.000553

|
| [3] |
Schwede T (2013) Protein modeling: What happened to the “protein structure gap”? Structure 21: 1531-1540. doi: 10.1016/j.str.2013.08.007
|
| [4] |
Torrisi M, Pollastri G, Le Q (2020) Deep learning methods in protein structure prediction. Comput Struct Biotechnol J 18: 1301-1310. doi: 10.1016/j.csbj.2019.12.011
|
| [5] |
Almeida JG, Preto AJ, Koukos PI, et al. (2017) Membrane proteins structures: A review on computational modeling tools. BBA Biomembr 1859: 2021-2039. doi: 10.1016/j.bbamem.2017.07.008
|
| [6] |
Korkmaz S, Duarte JM, Prlić A, et al. (2018) Investigation of protein quaternary structure via stoichiometry and symmetry information. PLoS One 13: e0197176. doi: 10.1371/journal.pone.0197176
|
| [7] |
Nealon JO, Philomina LS, McGuffin LJ (2017) Predictive and experimental approaches for elucidating protein–protein interactions and quaternary structures. Int J Mol Sci 18: 2623. doi: 10.3390/ijms18122623
|
| [8] |
Chowdhury B, Garai G (2017) A review on multiple sequence alignment from the perspective of genetic algorithm. Genomics 109: 419-431. doi: 10.1016/j.ygeno.2017.06.007
|
| [9] |
Saw AK, Tripathy BC, Nandi S (2019) Alignment-free similarity analysis for protein sequences based on fuzzy integral. Sci Rep 9: 2775. doi: 10.1038/s41598-019-39477-8
|
| [10] |
Cherstvy AG (2009) Positively charged residues in DNA-binding domains of structural proteins follow sequence-specific positions of DNA phosphate groups. J Phys Chem B 113: 4242-4247. doi: 10.1021/jp810009s
|
| [11] |
Moradpour D, Penin F (2013) Hepatitis C virus proteins: from structure to function. Hepatitis C Virus: from Molecular Virology to Antiviral Therapy Berlin: Springer, 113-142. doi: 10.1007/978-3-642-27340-7_5
|
| [12] | Paul D, Madan V, Ramirez O, et al. (2018) Glycine zipper motifs in hepatitis C virus nonstructural protein 4B are required for the establishment of viral replication organelles. J Virol 92: e01890-01817. |
| [13] |
Lundin M, Monné M, Widell A, et al. (2003) Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77: 5428-5438. doi: 10.1128/JVI.77.9.5428-5438.2003
|
| [14] |
Lundin M, Lindström H, Grönwall C, et al. (2006) Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J Gen Virol 87: 3263-3272. doi: 10.1099/vir.0.82211-0
|
| [15] |
Palomares-Jerez F, Nemesio H, Villalaín J (2012) The membrane spanning domains of protein NS4B from hepatitis C virus. BBA Biomembr 1818: 2958-2966. doi: 10.1016/j.bbamem.2012.07.022
|
| [16] |
Einav S, Elazar M, Danieli T, et al. (2004) A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J Virol 78: 11288-11295. doi: 10.1128/JVI.78.20.11288-11295.2004
|
| [17] |
Gouttenoire J, Penin F, Moradpour D (2010) Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20: 117-129. doi: 10.1002/rmv.640
|
| [18] |
Gouttenoire J, Montserret R, Paul D, et al. (2014) Aminoterminal amphipathic α-Helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog 10: e1004501. doi: 10.1371/journal.ppat.1004501
|
| [19] |
Yu GY, Lee KJ, Gao L, et al. (2006) Palmitoylation and polymerization of hepatitis C virus NS4B protein. J Virol 80: 6013-6023. doi: 10.1128/JVI.00053-06
|
| [20] |
Fogeron ML, Jirasko V, Penzel S, et al. (2016) Cell-free expression, purification, and membrane reconstitution for NMR studies of the nonstructural protein 4B from hepatitis C virus. J Biomol NMR 65: 87-98. doi: 10.1007/s10858-016-0040-2
|
| [21] |
Dobson L, Reményi I, Tusnády GE (2015) CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res 43: W408-W412. doi: 10.1093/nar/gkv451
|
| [22] |
Cserzö M, Wallin E, Simon I, et al. (1997) Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng 10: 673-676. doi: 10.1093/protein/10.6.673
|
| [23] |
Cserzo M, Eisenhaber F, Eisenhaber B, et al. (2004) TM or not TM: transmembrane protein prediction with low false positive rate using DAS-TMfilter. Bioinformatics 20: 136-137. doi: 10.1093/bioinformatics/btg394
|
| [24] |
Tusnády GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849-850. doi: 10.1093/bioinformatics/17.9.849
|
| [25] |
Shen H, Chou JJ (2008) MemBrain: improving the accuracy of predicting transmembrane helices. PLOS One 3: e2399. doi: 10.1371/journal.pone.0002399
|
| [26] |
Jones DT, Taylor WR, Thornton JM (1994) A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33: 3038-3049. doi: 10.1021/bi00176a037
|
| [27] |
Jones DT (2007) Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23: 538-544. doi: 10.1093/bioinformatics/btl677
|
| [28] |
Käll L, Krogh A, Sonnhammer ELL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338: 1027-1036. doi: 10.1016/j.jmb.2004.03.016
|
| [29] |
Daley DO, Rapp M, Granseth E, et al. (2005) Global topology analysis of the Escherichia coli inner membrane proteome. Science 308: 1321-1323. doi: 10.1126/science.1109730
|
| [30] |
Viklund H, Elofsson A (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24: 1662-1668. doi: 10.1093/bioinformatics/btn221
|
| [31] |
Reynolds SM, Käll L, Riffle ME, et al. (2008) Transmembrane topology and signal peptide prediction using dynamic bayesian networks. PLoS Comput Biol 4: e1000213. doi: 10.1371/journal.pcbi.1000213
|
| [32] |
Käll L, Krogh A, Sonnhammer ELL (2005) An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21: i251-i257. doi: 10.1093/bioinformatics/bti1014
|
| [33] |
Viklund H, Elofsson A (2004) Best α-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci 13: 1908-1917. doi: 10.1110/ps.04625404
|
| [34] |
Peters C, Tsirigos KD, Shu N, et al. (2016) Improved topology prediction using the terminal hydrophobic helices rule. Bioinformatics 32: 1158-1162. doi: 10.1093/bioinformatics/btv709
|
| [35] |
Juretić D, Zoranić L, Zucić D (2002) Basic charge clusters and prediction of membrane protein topology. J Chem Inf Comput Sci 42: 620-632. doi: 10.1021/ci010263s
|
| [36] |
Krogh A, Larsson B, Von Heijne G, et al. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305: 567-580. doi: 10.1006/jmbi.2000.4315
|
| [37] |
Kahsay RY, Gao G, Liao L (2005) An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 21: 1853-1858. doi: 10.1093/bioinformatics/bti303
|
| [38] | Hofmann K (1993) TMbase-A database of membrane spanning protein segments. Biol Chem 374: 166. |
| [39] |
Tsirigos KD, Peters C, Shu N, et al. (2015) The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 43: W401-W407. doi: 10.1093/nar/gkv485
|
| [40] |
Buchan DWA, Jones DT (2019) The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 47: W402-W407. doi: 10.1093/nar/gkz297
|
| [41] |
Hildebrand PW, Preissner R, Frömmel C (2004) Structural features of transmembrane helices. FEBS Lett 559: 145-151. doi: 10.1016/S0014-5793(04)00061-4
|
| [42] |
Saidijam M, Azizpour S, Patching SG (2018) Comprehensive analysis of the numbers, lengths and amino acid compositions of transmembrane helices in prokaryotic, eukaryotic and viral integral membrane proteins of high-resolution structure. J Biomol Struct Dyn 36: 443-464. doi: 10.1080/07391102.2017.1285725
|
| [43] |
Blum M, Chang HY, Chuguransky S, et al. (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49: D344-D354. doi: 10.1093/nar/gkaa977
|
| [44] |
Sigrist CJA, Cerutti L, Hulo N, et al. (2002) PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform 3: 265-274. doi: 10.1093/bib/3.3.265
|
| [45] |
Li S, Ye L, Yu X, et al. (2009) Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 391: 257-264. doi: 10.1016/j.virol.2009.06.039
|
| [46] |
Carrasco L (1978) Membrane leakiness after viral infection and a new approach to the development of antiviral agents. Nature 272: 694-699. doi: 10.1038/272694a0
|
| [47] |
Mehnert T, Routh A, Judge PJ, et al. (2008) Biophysical characterisation of Vpu from HIV-1 suggests a channel-pore dualism. Proteins 70: 1488-1497. doi: 10.1002/prot.21642
|
| [48] |
Lei J, Kusov Y, Hilgenfeld R (2018) Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res 149: 58-74. doi: 10.1016/j.antiviral.2017.11.001
|
| [49] |
Flock T, Venkatakrishnan AJ, Vinothkumar KR, et al. (2012) Deciphering membrane protein structures from protein sequences. Genome Biol 13: 160. doi: 10.1186/gb-2012-13-6-160
|
| [50] |
Cuthbertson JM, Doyle DA, Sansom MSP (2005) Transmembrane helix prediction: a comparative evaluation and analysis. Protein Eng Des Sel 18: 295-308. doi: 10.1093/protein/gzi032
|
| [51] |
Yan R, Xu D, Yang J, et al. (2013) A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep 3: 2619. doi: 10.1038/srep02619
|
| [52] |
Palomares-Jerez MF, Nemesio H, Franquelim HG, et al. (2013) N-terminal AH2 segment of protein NS4B from hepatitis C virus. Binding to and interaction with model biomembranes. BBA-Biomembr 1828: 1938-1952. doi: 10.1016/j.bbamem.2013.04.020
|
| [53] |
Illergård K, Ardell DH, Elofsson A (2009) Structure is three to ten times more conserved than sequence–a study of structural response in protein cores. Proteins 77: 499-508. doi: 10.1002/prot.22458
|
| [54] |
Sousounis K, Haney CE, Cao J, et al. (2012) Conservation of the three-dimensional structure in non-homologous or unrelated proteins. Hum Genomics 6: 1-10. doi: 10.1186/1479-7364-6-1
|
| [55] |
Doolittle RF (1981) Similar amino acid sequences: chance or common ancestry? Science 214: 149-159. doi: 10.1126/science.7280687
|
 biophy-08-02-013-s001.pdf biophy-08-02-013-s001.pdf |
 |
Figures(5) / Tables(2)

Ta-Chou Huang, Wolfgang B. Fischer. Sequence–function correlation of the transmembrane domains in NS4B of HCV using a computational approach[J]. AIMS Biophysics, 2021, 8(2): 165-181. doi: 10.3934/biophy.2021013







 DownLoad:
DownLoad: