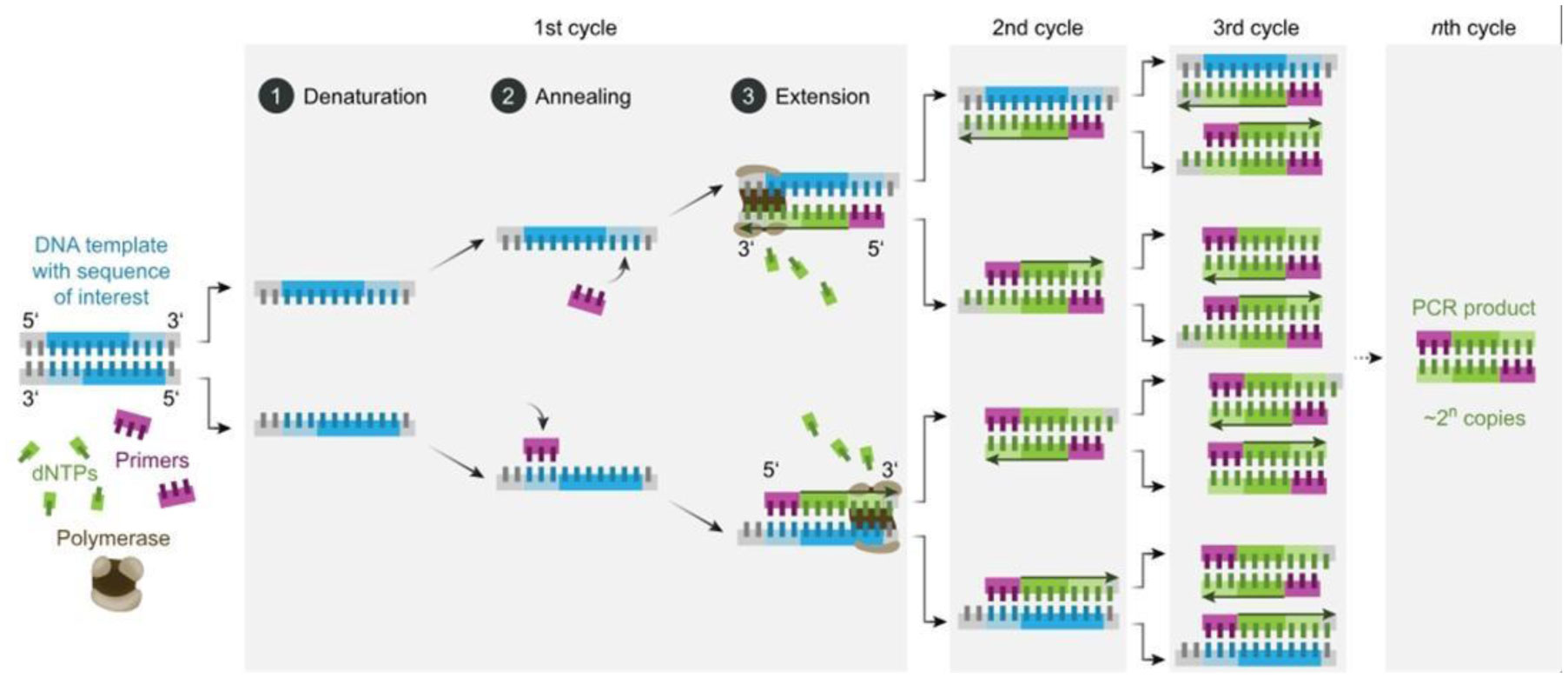
In humans particularly in children, adenovirus is one of the most common viruses that cause respiratory illnesses. Knowing how to detect adenovirus proficiently and rapidly will help reinforce surveillance of adenovirus infections, detect epidemic situations in real-time, and understand the trend of virus epidemics, which will allow effective actions to be taken quickly. The rapid detection of antiviral antibodies or viral antigens in clinical samples can be achieved by molecular diagnostic techniques like PCR, Real-Time PCR, LAMP, mPCR-RLB, PCR-ELISA, Tem-PCR, Gene Chip, and so on. Some of the molecular diagnostic methods are relatively economical, exceedingly sensitive and explicit. There are several commercially accessible molecular diagnostic techniques that enable their use in clinical laboratories all over the world. In this review, the principles, characteristics, and applications of molecular biology surveillance methods commonly used in labs and clinics for the detection of human adenoviruses are examined and highlighted.
Citation: Neelabh Datta. A review of molecular biology detection methods for human adenovirus[J]. AIMS Biophysics, 2023, 10(1): 95-120. doi: 10.3934/biophy.2023008
In humans particularly in children, adenovirus is one of the most common viruses that cause respiratory illnesses. Knowing how to detect adenovirus proficiently and rapidly will help reinforce surveillance of adenovirus infections, detect epidemic situations in real-time, and understand the trend of virus epidemics, which will allow effective actions to be taken quickly. The rapid detection of antiviral antibodies or viral antigens in clinical samples can be achieved by molecular diagnostic techniques like PCR, Real-Time PCR, LAMP, mPCR-RLB, PCR-ELISA, Tem-PCR, Gene Chip, and so on. Some of the molecular diagnostic methods are relatively economical, exceedingly sensitive and explicit. There are several commercially accessible molecular diagnostic techniques that enable their use in clinical laboratories all over the world. In this review, the principles, characteristics, and applications of molecular biology surveillance methods commonly used in labs and clinics for the detection of human adenoviruses are examined and highlighted.

| [1] |
Gallardo J, Pérez-Illana M, Martín-González N, et al. (2021) Adenovirus structure: what is new?. In J Mol Sci 22: 5240. https://doi.org/10.3390/ijms22105240 
|
| [2] |
Ebner K, Pinsker W, Lion T (2005) Comparative sequence analysis of the hexon gene in the entire spectrum of human adenovirus serotypes: phylogenetic, taxonomic, and clinical implications. J Virol 79: 12635-12642. https://doi.org/10.1128/JVI.79.20.12635-12642.2005
|
| [3] |
Suraka B, Usman U, Tijjani A (2022) A brief review on the molecular biology of human adenoviruses. Baghdad J Biochem Appl Biol Sci 3: 166-182. https://doi.org/10.47419/bjbabs.v3i03.146
|
| [4] |
Rowe WP, Huebner RJ, Gilmore LK, et al. (1953) Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Ex Biol Med 84: 570-573. https://doi.org/10.3181/00379727-84-20714
|
| [5] |
Lion T (2014) Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev 27: 441-462. https://doi.org/10.1128/CMR.00116-13
|
| [6] |
Hur SJ, Kim DH, Chun SC, et al. (2013) Effect of adenovirus and influenza virus infection on obesity. Life Sci 93: 531-535. https://doi.org/10.1016/j.lfs.2013.08.016
|
| [7] |
Mullis K, Faloona F, Scharf S, et al. (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51: 263-273. https://doi.org/10.1101/sqb.1986.051.01.032
|
| [8] |
Wu X, Zhang J, Lan W, et al. (2022) Molecular typing and rapid identification of human adenoviruses associated with respiratory diseases using universal PCR and sequencing primers for the three major capsid genes: penton base, hexon, and fiber. Front Microbiol 13: 911694. https://doi.org/10.3389/fmicb.2022.911694
|
| [9] |
Shieh WJ (2022) Human adenovirus infections in pediatric population–an update on clinico-pathologic correlation. Biomed J 45: 38-49. https://doi.org/10.1016/j.bj.2021.08.009to
|
| [10] |
Pehler-Harrington K, Khanna M, Waters CR, et al. (2004) Rapid detection and identification of human adenovirus species by adenoplex, a multiplex PCR-enzyme hybridization assay. J Clin Microbiol 42: 4072-4076. https://doi.org/10.1128/JCM.42.9.4072-4076.2004
|
| [11] |
Al-Siyabi T, Binkhamis K, Wilcox M, et al. (2013) A cost effective real-time PCR for the detection of adenovirus from viral swabs. Virol J 10: 184. https://doi.org/10.1186/1743-422X-10-184
|
| [12] |
Casas I, Avellon A, Mosquera M, et al. (2005) Molecular identification of adenoviruses in clinical samples by analyzing a partial hexon genomic region. J Clin Microbiol 43: 6176-6182. https://doi.org/10.1128/JCM.43.12.6176-6182.2005
|
| [13] |
He JW, Jiang S (2005) Quantification of enterococci and human adenoviruses in environmental samples by real-time PCR. Appl Environ Microbio 71: 2250-2255. https://doi.org/10.1128/AEM.71.5.2250-2255.2005
|
| [14] |
Dolskiy AA, Grishchenko IV, Yudkin DV (2020) Cell cultures for virology: usability, advantages, and prospects. Int J Mo Sci 21: 7978. https://doi.org/10.3390/ijms21217978
|
| [15] |
Shike H, Shimizu C, Kanegaye J, et al. (2005) Quantitation of adenovirus genome during acute infection in normal children. Pediatr Infect Dis J 24: 29-33. https://doi.org/10.1097/01.inf.0000148882.97399.79
|
| [16] |
Choi S, Jiang SC (2005) Real-time PCR quantification of human adenoviruses in urban rivers indicates genome prevalence but low infectivity. Appl Enviro Microb 71: 7426-7433. https://doi.org/10.1128/AEM.71.11.7426-7433.2005
|
| [17] |
Morozumi M, Shimizu H, Matsushima Y, et al. (2014) Evaluation of new immunochromatographic assay kit for adenovirus detection in throat swab: Comparison with culture and real-time PCR results. J Infect Chemother 20: 303-306. https://doi.org/10.1016/j.jiac.2014.01.005
|
| [18] |
Jones MS, Hudson NR, Gibbins C, et al. (2011) Evaluation of type-specific real-time PCR assays using the LightCycler and J.B.A.I.D.S. for detection of adenoviruses in species HAdV-C. PloS One 6: e26862. https://doi.org/10.1371/journal.pone.0026862
|
| [19] |
Feeney SA, Armstrong VJ, Mitchell SJ, et al. (2011) Development and clinical validation of multiplex TaqMan® assays for rapid diagnosis of viral gastroenteritis. J Med Virol 83: 1650-1656. https://doi.org/10.1002/jmv.22162
|
| [20] | Li F, Zhao LQ, Deng J, et al. (2013) Detecting human adenoviruses in respiratory samples collected from children with acute respiratory infections by loop-mediated isothermal amplification. Chinese J Pediatr 51: 52-57. |
| [21] |
Metzger-Boddien C, Kehle J (2005) Development and evaluation of a sensitive PCR-ELISA for detection of adenoviruses in feces. Intervirology 48: 297-300. https://doi.org/10.1159/000085098
|
| [22] | Murali S, Langston AA, Nolte FS, et al. (2009) Detection of respiratory viruses with a multiplex polymerase chain reaction assay (multicode-PLx respiratory virus panel) in patients with hematologic malignancies. Leukemia & lymphoma 50: 619-624. https://doi.org/10.1080/10428190902777665 |
| [23] | van Maarseveen NM, Wessels E, de Brouwer CS, et al. (2010) Diagnosis of viral gastroenteritis by simultaneous detection of adenovirus group F, astrovirus, rotavirus group A, norovirus genogroups I and II, and sapovirus in two internally controlled multiplex real-time PCR assays. J Clin Microbiol J Clin Microbiol 49: 205-210. https://doi.org/10.1016/j.jcv.2010.07.019 |
| [24] |
Lalli MA, Langmade JS, Chen X, et al. (2021) Rapid and extraction-free detection of SARS-CoV-2 from saliva by colorimetric reverse-transcription loop-mediated isothermal amplification. Clin Chem 67: 415-424. https://doi.org/10.1093/clinchem/hvaa267
|
| [25] | Foo PC, Nurul Najian AB, Muhamad NA, et al. (2020) Loop-mediated isothermal amplification (LAMP) reaction as viable PCR substitute for diagnostic applications: a comparative analysis study of LAMP, conventional PCR, nested PCR (nPCR) and real-time PCR (qPCR) based on Entamoeba histolytica DNA derived from faecal sample. BMC Biotechnol 20–34. https://doi.org/10.1186/s12896-020-00629-8 |
| [26] |
Wang Y, Kong F, Yang Y, et al. (2008) A multiplex PCR-based reverse line blot hybridization (mPCR/RLB) assay for detection of bacterial respiratory pathogens in children with pneumonia. Pediatr Pulm 43: 150-159. https://doi.org/10.1002/ppul.20749
|
| [27] | O'Sullivan MV, Zhou F, Sintchenko V, et al. (2011) Multiplex PCR and reverse line blot hybridization assay (mPCR/RLB). J Vis Exp 54: e2781. https://doi.org/10.3791/2781 |
| [28] |
Pierce VM, Elkan M, Leet M, et al. (2012) Comparison of the Idaho technology film array system to real-time PCR for detection of respiratory pathogens in children. J Clin Microbiol 50: 364-371. https://doi.org/10.1128/JCM.05996-11
|
| [29] | Zink S, Cirino NM, Egan C (2011) Multiplex PCR product detection and discrimination. Mol Microbiol: Diagn Prin Pract 325–341. https://doi.org/10.1128/9781555816834.ch21 |
| [30] | Silbereisen A Monoclonal antibodies as tools in antigen detection assay and vaccine development: design of a sensitive detection test for Brucella bacteria and profiling of the malaria vaccine candidate antigen reticulocyte-binding homolog 2 (PfRH2), University_of_Basel (2015). |
| [31] |
Puppe W, Weigl JA, Aron G, et al. (2004) Evaluation of a multiplex reverse transcriptase PCR ELISA for the detection of nine respiratory tract pathogens. J Clin Virol 30: 165-174. https://doi.org/10.1016/j.jcv.2003.10.003
|
| [32] |
Puppe W, Weigl J, Gröndahl B, et al. (2013) Validation of a multiplex reverse transcriptase PCR ELISA for the detection of 19 respiratory tract pathogens. Infection 41: 77-91. https://doi.org/10.1007/s15010-012-0298-6
|
| [33] | Zulauf BJ Multiplex real-time PCR in the detection and differentiation of bovine respiratory disease pathogens (2007). Available from: https://ir.library.oregonstate.edu/concern/graduate_thesis_or_dissertations/0g354j20f |
| [34] | Zhang Y, Cao L, Xu Z, et al. (2020) Evaluation of a multiplex PCR assay for detection of respiratory viruses and Mycoplasma pneumoniae in oropharyngeal swab samples from outpatients. J Clin Lab Anal 34: e23032. https://doi.org/10.1002/jcla.23032 |
| [35] |
Lee JA, Kim NH, Kim SJ, et al. (2005) Rapid identification of human adenovirus types 3 and 7 from respiratory specimens via multiplex type-specific PCR. J Clin Microbiol 43: 5509-5514. https://doi.org/10.1128/JCM.43.11.5509-5514.2005
|
| [36] |
Chin EL, da Silva C, Hegde M (2013) Assessment of clinical analytical sensitivity and specificity of next-generation sequencing for detection of simple and complex mutations. BMC Genet 14: 6. https://doi.org/10.1186/1471-2156-14-6
|
| [37] |
Reijans M, Dingemans G, Klaassen CH, et al. (2008) RespiFinder: a new multiparameter test to differentially identify fifteen respiratory viruses. J Clin Microbiol 46: 1232-1240. https://doi.org/10.1128/JCM.02294-07
|
| [38] |
Thevendran R, Citartan M (2022) Assays to estimate the binding affinity of aptamers. Talanta 238: 122971. https://doi.org/10.1016/j.talanta.2021.122971
|
| [39] |
Shaffer SM, Joshi RP, Chambers BS, et al. (2015) Multiplexed detection of viral infections using rapid in situ RNA analysis on a chip. Lab Chip 15: 3170-3182. https://doi.org/10.1039/c5lc00459d
|
| [40] |
Renois F, Talmud D, Huguenin A, et al. (2010) Rapid detection of respiratory tract viral infections and coinfections in patients with influenza-like illnesses by use of reverse transcription-PCR DNA microarray systems. J Clin Microbiol 48: 3836-3842. https://doi.org/10.1128/JCM.00733-10
|
| [41] |
Daly GM, Leggett RM, Rowe W, et al. (2015) Host subtraction, filtering and assembly validations for novel viral discovery using next generation sequencing data. PloS one 10: e0129059. https://doi.org/10.1371/journal.pone.0129059
|
| [42] |
Fujii H, Kakiuchi S, Tsuji M, et al. (2018) Application of next-generation sequencing to detect acyclovir-resistant herpes simplex virus type 1 variants at low frequency in thymidine kinase gene of the isolates recovered from patients with hematopoietic stem cell transplantation. J Virol Methods 251: 123-128. https://doi.org/10.1016/j.jviromet.2017.10.019
|
| [43] |
Zhang W, Ehrhardt A (2017) Getting genetic access to natural adenovirus genomes to explore vector diversity. Virus genes 53: 675-683. https://doi.org/10.1007/s11262-017-1487-2
|
| [44] |
Capobianchi MR, Giombini E, Rozera G (2013) Next-generation sequencing technology in clinical virology. Clin Microbiol Infec 19: 15-22. https://doi.org/10.1111/1469-0691.12056
|
| [45] | Reta DH, Tessema TS, Ashenef AS, et al. (2020) Molecular and immunological ddiagnostic ttechniques of medical viruses. Int J Microbiol 2020: 8832728. https://doi.org/10.1155/2020/8832728 |
| [46] |
Salez N, Vabret A, Leruez-Ville M, et al. (2015) Evaluation of four commercial multiplex molecular tests for the diagnosis of acute respiratory infections. PloS One 10: e0130378. https://doi.org/10.1371/journal.pone.0130378
|








Figures(9)

Neelabh Datta. A review of molecular biology detection methods for human adenovirus[J]. AIMS Biophysics, 2023, 10(1): 95-120. doi: 10.3934/biophy.2023008







 DownLoad:
DownLoad: