Citation: Marcos Pérez-Losada, Keith A. Crandall, Robert J. Freishtat. Comparison of two commercial DNA extraction kits for the analysis of nasopharyngeal bacterial communities[J]. AIMS Microbiology, 2016, 2(2): 108-119. doi: 10.3934/microbiol.2016.2.108

| [1] |
Ding T, Schloss PD (2014) Dynamics and associations of microbial community types across the human body. Nature 509: 357–360. doi: 10.1038/nature13178

|
| [2] |
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. doi: 10.1038/nature11234
|
| [3] |
Human Microbiome Project C (2012) A framework for human microbiome research. Nature 486: 215–221. doi: 10.1038/nature11209
|
| [4] |
Integrative HMPRNC (2014) The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 16: 276–289. doi: 10.1016/j.chom.2014.08.014
|
| [5] |
Marchesi JR, Ravel J (2015) The vocabulary of microbiome research: a proposal. Microbiome 3: 31. doi: 10.1186/s40168-015-0094-5
|
| [6] | Kuczynski J, Lauber CL, Walters WA, et al. (2012) Experimental and analytical tools for studying the human microbiome. Nat Rev Genet 13: 47–58. |
| [7] | Althani AA, Marei HE, Hamdi WS, et al. (2015) Human Microbiome and its Association With Health and Diseases. J Cell Physiol 231: 1688–1694. |
| [8] |
Martin R, Miquel S, Langella P, et al. (2014) The role of metagenomics in understanding the human microbiome in health and disease. Virulence 5: 413–423. doi: 10.4161/viru.27864
|
| [9] |
Cox MJ, Cookson WO, Moffatt MF (2013) Sequencing the human microbiome in health and disease. Hum Mol Genet 22: R88–94. doi: 10.1093/hmg/ddt398
|
| [10] |
Brooks JP, Edwards DJ, Harwich MD, Jr., et al. (2015) The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15: 66. doi: 10.1186/s12866-015-0351-6
|
| [11] | Abusleme L, Hong BY, Dupuy AK, et al. (2014) Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J Oral Microbiol 6. |
| [12] |
Wu GD, Lewis JD, Hoffmann C, et al. (2010) Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol 10: 206. doi: 10.1186/1471-2180-10-206
|
| [13] |
Momozawa Y, Deffontaine V, Louis E, et al. (2011) Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PLOS ONE 6: e16952. doi: 10.1371/journal.pone.0016952
|
| [14] |
Lazarevic V, Gaia N, Girard M, et al. (2013) Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLOS ONE 8: e67699. doi: 10.1371/journal.pone.0067699
|
| [15] |
Willner D, Daly J, Whiley D, et al. (2012) Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLOS ONE 7: e34605. doi: 10.1371/journal.pone.0034605
|
| [16] |
Bogaert D, De Groot R, Hermans PW (2004) Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4: 144–154. doi: 10.1016/S1473-3099(04)00938-7
|
| [17] | Garcia-Rodriguez JA, Fresnadillo Martinez MJ (2002) Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J Antimicrob Chemother 50 Suppl S2: 59–73. |
| [18] |
Biesbroek G, Tsivtsivadze E, Sanders EA, et al. (2014) Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am J Respir Crit Care Med 190: 1283–1292. doi: 10.1164/rccm.201407-1240OC
|
| [19] |
Teo SM, Mok D, Pham K, et al. (2015) The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17: 704–715. doi: 10.1016/j.chom.2015.03.008
|
| [20] |
Feazel LM, Santorico SA, Robertson CE, et al. (2015) Effects of Vaccination with 10-Valent Pneumococcal Non-Typeable Haemophilus influenza Protein D Conjugate Vaccine (PHiD-CV) on the Nasopharyngeal Microbiome of Kenyan Toddlers. PLOS ONE 10: e0128064. doi: 10.1371/journal.pone.0128064
|
| [21] | Prevaes SM, de Winter-de Groot KM, Janssens HM, et al. (2015) Development of the Nasopharyngeal Microbiota in Infants with Cystic Fibrosis. Am J Respir Crit Care Med. |
| [22] |
Cremers AJ, Zomer AL, Gritzfeld JF, et al. (2014) The adult nasopharyngeal microbiome as a determinant of pneumococcal acquisition. Microbiome 2: 44. doi: 10.1186/2049-2618-2-44
|
| [23] |
Allen EK, Koeppel AF, Hendley JO, et al. (2014) Characterization of the nasopharyngeal microbiota in health and during rhinovirus challenge. Microbiome 2: 22. doi: 10.1186/2049-2618-2-22
|
| [24] | Biesbroek G, Bosch AA, Wang X, et al. (2014) The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am J Respir Crit Care Med 190: 298–308. |
| [25] |
Sakwinska O, Bastic Schmid V, Berger B, et al. (2014) Nasopharyngeal microbiota in healthy children and pneumonia patients. J Clin Microbiol 52: 1590–1594. doi: 10.1128/JCM.03280-13
|
| [26] |
Bassis CM, Tang AL, Young VB, et al. (2014) The nasal cavity microbiota of healthy adults. Microbiome 2: 27. doi: 10.1186/2049-2618-2-27
|
| [27] |
Perez-Losada M, Castro-Nallar E, Bendall ML, et al. (2015) Dual Transcriptomic Profiling of Host and Microbiota during Health and Disease in Pediatric Asthma. PLOS ONE 10: e0131819. doi: 10.1371/journal.pone.0131819
|
| [28] |
Castro-Nallar E, Shen Y, Freishtat RJ, et al. (2015) Integrating metagenomics and host gene expression to characterize asthma-associated microbial communities. BMC Medical Genomics 8: 50. doi: 10.1186/s12920-015-0121-1
|
| [29] |
Bogaert D, Keijser B, Huse S, et al. (2011) Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLOS ONE 6: e17035. doi: 10.1371/journal.pone.0017035
|
| [30] | Pérez-Losada M, Crandall KA, Freishtat RJ (2016) Two sampling methods yield distinct microbial signatures in the nasopharynx of asthmatic children. Microbiome[in press]. |
| [31] |
Benton AS, Wang Z, Lerner J, et al. (2010) Overcoming heterogeneity in pediatric asthma: tobacco smoke and asthma characteristics within phenotypic clusters in an African American cohort. J Asthma 47: 728–734. doi: 10.3109/02770903.2010.491142
|
| [32] |
Kozich JJ, Westcott SL, Baxter NT, et al. (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79: 5112–5120. doi: 10.1128/AEM.01043-13
|
| [33] |
Schloss PD, Westcott SL, Ryabin T, et al. (2009) Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl Environ Microbiol 75: 7537–7541. doi: 10.1128/AEM.01541-09
|
| [34] |
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLOS ONE 6: e27310. doi: 10.1371/journal.pone.0027310
|
| [35] |
Edgar RC, Haas BJ, Clemente JC, et al. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200. doi: 10.1093/bioinformatics/btr381
|
| [36] |
Wang Q, Garrity GM, Tiedje JM, et al. (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267. doi: 10.1128/AEM.00062-07
|
| [37] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. doi: 10.1038/nmeth.f.303
|
| [38] | Price MN, Dehal PS, Arkin AP (2010) FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 5. |
| [39] |
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Conserv 61: 1–10. doi: 10.1016/0006-3207(92)91201-3
|
| [40] |
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14: 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
|
| [41] | RStudioTeam (2015) RStudio: Integrated Development for R. RStudio, Inc., Boston, MA URL http://www.rstudio.com/. |
| [42] |
Biesbroek G, Wang X, Keijser BJ, et al. (2014) Seven-valent pneumococcal conjugate vaccine and nasopharyngeal microbiota in healthy children. Emerg Infect Dis 20: 201–210. doi: 10.3201/eid2002.131220
|
| [43] |
Yan M, Pamp SJ, Fukuyama J, et al. (2013) Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe 14: 631–640. doi: 10.1016/j.chom.2013.11.005
|
| [44] | Cremers AJH, Zomer AL, Gritzfeld JF, et al. (2014) The adult nasopharyngeal microbiome as a determinant of pneumococcal acquisition. Microbiome 2. |
| [45] | Bassis CM, Erb-Downward JR, Dickson RP, et al. (2015) Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio 6: e00037. |
| [46] |
Morgan JL, Darling AE, Eisen JA (2010) Metagenomic sequencing of an in vitro-simulated microbial community. PLOS ONE 5: e10209. doi: 10.1371/journal.pone.0010209
|
| [47] |
Yuan S, Cohen DB, Ravel J, et al. (2012) Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLOS ONE 7: e33865. doi: 10.1371/journal.pone.0033865
|
| [48] |
P OC, Aguirre de Carcer D, Jones M, et al. (2011) The effects from DNA extraction methods on the evaluation of microbial diversity associated with human colonic tissue. Microb Ecol 61: 353–362. doi: 10.1007/s00248-010-9771-x
|
| [49] | Mackenzie BW, Waite DW, Taylor MW (2015) Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front Microbiol 6: 130. |
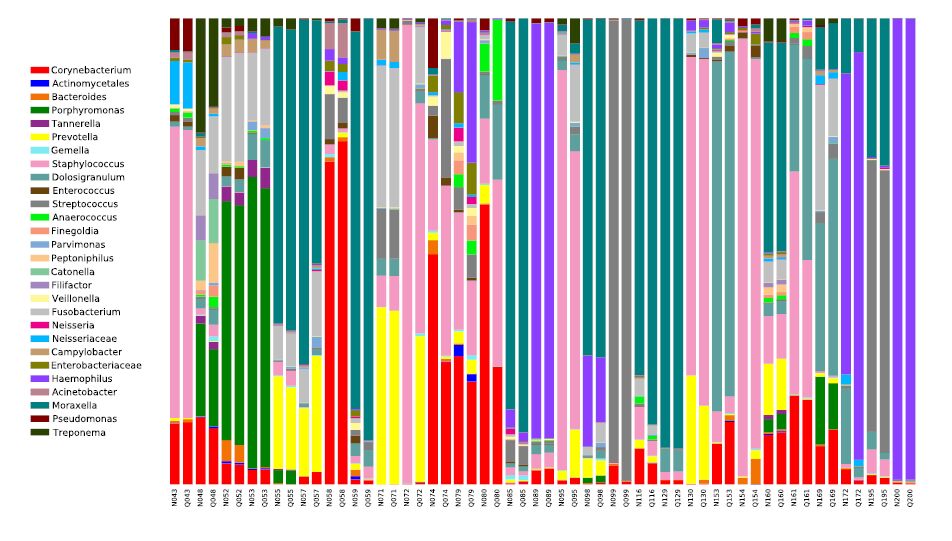
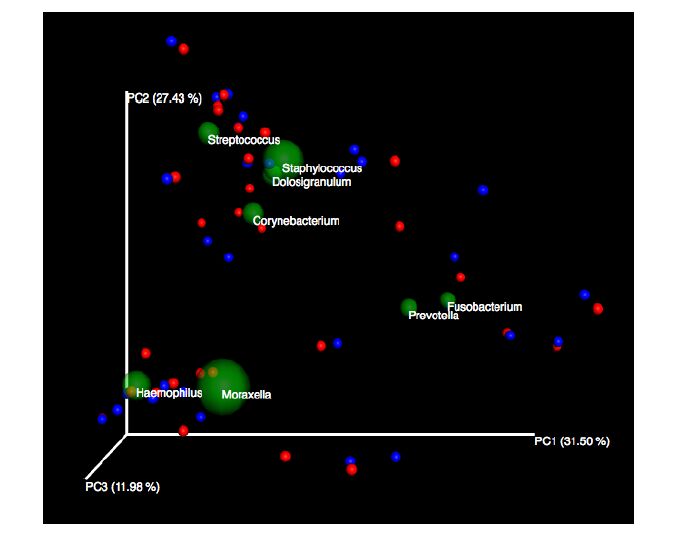
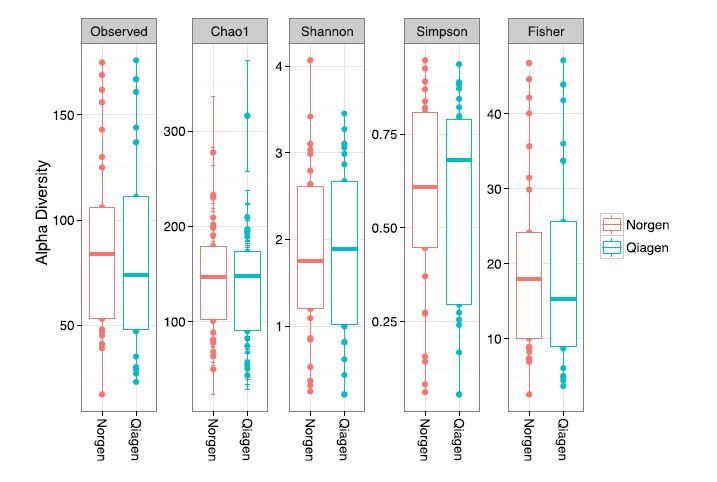

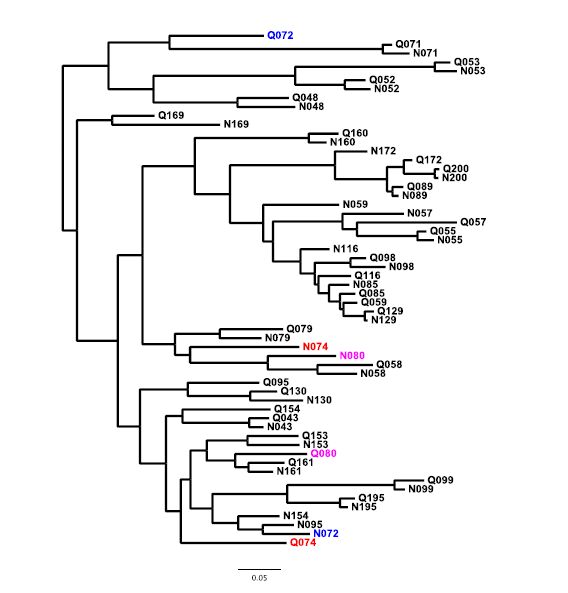
Figures(5)

Marcos Pérez-Losada, Keith A. Crandall, Robert J. Freishtat. Comparison of two commercial DNA extraction kits for the analysis of nasopharyngeal bacterial communities[J]. AIMS Microbiology, 2016, 2(2): 108-119. doi: 10.3934/microbiol.2016.2.108







 DownLoad:
DownLoad: