Citation: Jack C. Leo, Dirk Linke. A unified model for BAM function that takes into account type Vc secretion and species differences in BAM composition[J]. AIMS Microbiology, 2018, 4(3): 455-468. doi: 10.3934/microbiol.2018.3.455

| [1] |
Klauser T, Pohlner J, Meyer TF (1993) The secretion pathway of IgA protease-type proteins in Gram-negative bacteria. Bioessays 15: 799–805. doi: 10.1002/bies.950151205

|
| [2] |
Pohlner J, Halter R, Beyreuther K, et al. (1987) Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325: 458–462. doi: 10.1038/325458a0
|
| [3] | Klauser T, Pohlner J, Meyer TF (1990) Extracellular transport of cholera toxin B subunit using Neisseria IgA protease beta-domain: conformation-dependent outer membrane translocation. EMBO J 9: 1991–1999. |
| [4] |
Nicolay T, Vanderleyden J, Spaepen S (2015) Autotransporter-based cell surface display in Gram-negative bacteria. Crit Rev Microbiol 41: 109–123. doi: 10.3109/1040841X.2013.804032
|
| [5] |
Henderson IR, Navarro-Garcia F, Desvaux M, et al. (2004) Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev 68: 692–744. doi: 10.1128/MMBR.68.4.692-744.2004
|
| [6] | Fan E, Chauhan N, Udatha DB, et al. (2016) Type V secretion systems in bacteria. Microbiol Spectr 4. |
| [7] |
Guerin J, Bigot S, Schneider R, et al. (2017) Two-partner secretion: combining efficiency and simplicity in the secretion of large proteins for bacteria-host and bacteria-bacteria interactions. Front Cell Infect Mi 7: 148. doi: 10.3389/fcimb.2017.00148
|
| [8] |
Bassler J, Alvarez BH, Hartmann MD, et al. (2015) A domain dictionary of trimeric autotransporter adhesins. Int J Med Microbiol 305: 265–275. doi: 10.1016/j.ijmm.2014.12.010
|
| [9] |
Linke D, Riess T, Autenrieth IB, et al. (2006) Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol 14: 264–270. doi: 10.1016/j.tim.2006.04.005
|
| [10] | Salacha R, Kovacic F, Brochier-Armanet C, et al. (2010) The Pseudomonas aeruginosa patatin-like protein PlpD is the archetype of a novel Type V secretion system. Environ Microbiol 12: 1498–1512. |
| [11] |
Casasanta MA, Yoo CC, Smith HB, et al. (2017) A chemical and biological toolbox for Type Vd secretion: Characterization of the phospholipase A1 autotransporter FplA from Fusobacterium nucleatum. J Biol Chem 292: 20240–20254. doi: 10.1074/jbc.M117.819144
|
| [12] |
Leo JC, Oberhettinger P, Schutz M, et al. (2015) The inverse autotransporter family: intimin, invasin and related proteins. Int J Med Microbiol 305: 276–282. doi: 10.1016/j.ijmm.2014.12.011
|
| [13] |
Wu T, Malinverni J, Ruiz N, et al. (2005) Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121: 235–245. doi: 10.1016/j.cell.2005.02.015
|
| [14] |
Knowles TJ, Scott-Tucker A, Overduin M, et al. (2009) Membrane protein architects: the role of the BAM complex in outer membrane protein assembly. Nat Rev Microbiol 7: 206–214. doi: 10.1038/nrmicro2069
|
| [15] |
Malinverni JC, Werner J, Kim S, et al. (2006) YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61: 151–164. doi: 10.1111/j.1365-2958.2006.05211.x
|
| [16] |
Sklar JG, Wu T, Gronenberg LS, et al. (2007) Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. P Natl Acad Sci USA 104: 6400–6405. doi: 10.1073/pnas.0701579104
|
| [17] |
Robert V, Volokhina EB, Senf F, et al. (2006) Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Biol 4: e377. doi: 10.1371/journal.pbio.0040377
|
| [18] |
Noinaj N, Kuszak AJ, Gumbart JC, et al. (2013) Structural insight into the biogenesis of beta-barrel membrane proteins. Nature 501: 385–390. doi: 10.1038/nature12521
|
| [19] |
Gu Y, Li H, Dong H, et al. (2016) Structural basis of outer membrane protein insertion by the BAM complex. Nature 531: 64–69. doi: 10.1038/nature17199
|
| [20] |
Han L, Zheng J, Wang Y, et al. (2016) Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol 23: 192–196. doi: 10.1038/nsmb.3181
|
| [21] |
Albrecht R, Schutz M, Oberhettinger P, et al. (2014) Structure of BamA, an essential factor in outer membrane protein biogenesis. Acta Crystallogr D Biol Crystallogr 70: 1779–1789. doi: 10.1107/S1399004714007482
|
| [22] |
Bakelar J, Buchanan SK, Noinaj N (2016) The structure of the beta-barrel assembly machinery complex. Science 351: 180–186. doi: 10.1126/science.aad3460
|
| [23] |
Iadanza MG, Higgins AJ, Schiffrin B, et al. (2016) Lateral opening in the intact beta-barrel assembly machinery captured by cryo-EM. Nat Commun 7: 12865. doi: 10.1038/ncomms12865
|
| [24] |
Noinaj N, Kuszak AJ, Balusek C, et al. (2014) Lateral opening and exit pore formation are required for BamA function. Structure 22: 1055–1062. doi: 10.1016/j.str.2014.05.008
|
| [25] |
Gatzeva-Topalova PZ, Warner LR, Pardi A, et al. (2010) Structure and flexibility of the complete periplasmic domain of BamA: the protein insertion machine of the outer membrane. Structure 18: 1492–1501. doi: 10.1016/j.str.2010.08.012
|
| [26] |
Gatzeva-Topalova PZ, Walton TA, Sousa MC (2008) Crystal structure of YaeT: conformational flexibility and substrate recognition. Structure 16: 1873–1881. doi: 10.1016/j.str.2008.09.014
|
| [27] |
Knowles TJ, Jeeves M, Bobat S, et al. (2008) Fold and function of polypeptide transport-associated domains responsible for delivering unfolded proteins to membranes. Mol Microbiol 68: 1216–1227. doi: 10.1111/j.1365-2958.2008.06225.x
|
| [28] |
Lee J, Xue M, Wzorek JS, et al. (2016) Characterization of a stalled complex on the beta-barrel assembly machine. P Natl Acad Sci USA 113: 8717–8722. doi: 10.1073/pnas.1604100113
|
| [29] |
Schiffrin B, Calabrese AN, Higgins AJ, et al. (2017) Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J Mol Biol 429: 3776–3792. doi: 10.1016/j.jmb.2017.09.008
|
| [30] | Hohr AIC, Lindau C, Wirth C, et al. (2018) Membrane protein insertion through a mitochondrial beta-barrel gate. Science 359. |
| [31] |
Jain S, Goldberg MB (2007) Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J Bacteriol 189: 5393–5398. doi: 10.1128/JB.00228-07
|
| [32] |
Sauri A, Soprova Z, Wickstrom D, et al. (2009) The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155: 3982–3991. doi: 10.1099/mic.0.034991-0
|
| [33] |
Lehr U, Schutz M, Oberhettinger P, et al. (2010) C-terminal amino acid residues of the trimeric autotransporter adhesin YadA of Yersinia enterocolitica are decisive for its recognition and assembly by BamA. Mol Microbiol 78: 932–946. doi: 10.1111/j.1365-2958.2010.07377.x
|
| [34] |
Oberhettinger P, Leo JC, Linke D, et al. (2015) The inverse autotransporter intimin exports its passenger domain via a hairpin intermediate. J Biol Chem 290: 1837–1849. doi: 10.1074/jbc.M114.604769
|
| [35] |
Albenne C, Ieva R (2017) Job contenders: roles of the beta-barrel assembly machinery and the translocation and assembly module in autotransporter secretion. Mol Microbiol 106: 505–517. doi: 10.1111/mmi.13832
|
| [36] |
Pavlova O, Peterson JH, Ieva R, et al. (2013) Mechanistic link between beta barrel assembly and the initiation of autotransporter secretion. P Natl Acad Sci USA 110: E938–E947. doi: 10.1073/pnas.1219076110
|
| [37] |
Noinaj N, Gumbart JC, Buchanan SK (2017) The beta-barrel assembly machinery in motion. Nat Rev Microbiol 15: 197–204. doi: 10.1038/nrmicro.2016.191
|
| [38] |
Arnold T, Zeth K, Linke D (2010) Omp85 from the thermophilic cyanobacterium Thermosynechococcus elongatus differs from proteobacterial Omp85 in structure and domain composition. J Biol Chem 285: 18003–18015. doi: 10.1074/jbc.M110.112516
|
| [39] |
Koenig P, Mirus O, Haarmann R, et al. (2010) Conserved properties of polypeptide transport-associated (POTRA) domains derived from cyanobacterial Omp85. J Biol Chem 285: 18016–18024. doi: 10.1074/jbc.M110.112649
|
| [40] |
Bos MP, Grijpstra J, Tommassen-van Boxtel R, et al. (2014) Involvement of Neisseria meningitidis lipoprotein GNA2091 in the assembly of a subset of outer membrane proteins. J Biol Chem 289: 15602–15610. doi: 10.1074/jbc.M113.539510
|
| [41] |
Volokhina EB, Beckers F, Tommassen J, et al. (2009) The beta-barrel outer membrane protein assembly complex of Neisseria meningitidis. J Bacteriol 191: 7074–7085. doi: 10.1128/JB.00737-09
|
| [42] |
Anwari K, Webb CT, Poggio S, et al. (2012) The evolution of new lipoprotein subunits of the bacterial outer membrane BAM complex. Mol Microbiol 84: 832–844. doi: 10.1111/j.1365-2958.2012.08059.x
|
| [43] |
Paramasivam N, Habeck M, Linke D (2012) Is the C-terminal insertional signal in Gram-negative bacterial outer membrane proteins species-specific or not? BMC Genomics 13: 510. doi: 10.1186/1471-2164-13-510
|
| [44] |
Volokhina EB, Grijpstra J, Beckers F, et al. (2013) Species-specificity of the BamA component of the bacterial outer membrane protein-assembly machinery. PLoS One 8: e85799. doi: 10.1371/journal.pone.0085799
|
| [45] |
Webb CT, Heinz E, Lithgow T (2012) Evolution of the beta-barrel assembly machinery. Trends Microbiol 20: 612–620. doi: 10.1016/j.tim.2012.08.006
|
| [46] |
Iqbal H, Kenedy MR, Lybecker M, et al. (2016) The TamB ortholog of Borrelia burgdorferi interacts with the beta-barrel assembly machine (BAM) complex protein BamA. Mol Microbiol 102: 757–774. doi: 10.1111/mmi.13492
|
| [47] |
Selkrig J, Mosbahi K, Webb CT, et al. (2012) Discovery of an archetypal protein transport system in bacterial outer membranes. Nat Struct Mol Biol 19: 506–510. doi: 10.1038/nsmb.2261
|
| [48] |
Gruss F, Zahringer F, Jakob RP, et al. (2013) The structural basis of autotransporter translocation by TamA. Nat Struct Mol Biol 20: 1318–1320. doi: 10.1038/nsmb.2689
|
| [49] |
Josts I, Stubenrauch CJ, Vadlamani G, et al. (2017) The structure of a conserved domain of TamB reveals a hydrophobic beta taco fold. Structure 25: 1898–1906. doi: 10.1016/j.str.2017.10.002
|
| [50] |
Shen HH, Leyton DL, Shiota T, et al. (2014) Reconstitution of a nanomachine driving the assembly of proteins into bacterial outer membranes. Nat Commun 5: 5078. doi: 10.1038/ncomms6078
|
| [51] |
Stubenrauch C, Belousoff MJ, Hay ID, et al. (2016) Effective assembly of fimbriae in Escherichia coli depends on the translocation assembly module nanomachine. Nat Microbiol 1: 16064. doi: 10.1038/nmicrobiol.2016.64
|
| [52] |
Kang'ethe W, Bernstein HD (2013) Charge-dependent secretion of an intrinsically disordered protein via the autotransporter pathway. P Natl Acad Sci USA 110: E4246–E4255. doi: 10.1073/pnas.1310345110
|
| [53] |
Norell D, Heuck A, Tran-Thi TA, et al. (2014) Versatile in vitro system to study translocation and functional integration of bacterial outer membrane proteins. Nat Commun 5: 5396. doi: 10.1038/ncomms6396
|
| [54] |
Heinz E, Stubenrauch CJ, Grinter R, et al. (2016) Conserved features in the structure, mechanism, and biogenesis of the inverse autotransporter protein family. Genome Biol Evol 8: 1690–1705. doi: 10.1093/gbe/evw112
|
| [55] | Heinz E, Lithgow T (2014) A comprehensive analysis of the Omp85/TpsB protein superfamily structural diversity, taxonomic occurrence, and evolution. Front Microbiol 5: 370. |
| [56] |
Heinz E, Selkrig J, Belousoff MJ, et al. (2015) Evolution of the Translocation and Assembly Module (TAM). Genome Biol Evol 7: 1628–1643. doi: 10.1093/gbe/evv097
|
| [57] |
Remmert M, Biegert A, Linke D, et al. (2010) Evolution of outer membrane beta-barrels from an ancestral beta beta hairpin. Mol Biol Evol 27: 1348–1358. doi: 10.1093/molbev/msq017
|
| [58] |
Remmert M, Linke D, Lupas AN, et al. (2009) HHomp-prediction and classification of outer membrane proteins. Nucleic Acids Res 37: W446–W451. doi: 10.1093/nar/gkp325
|
| [59] |
Kleinschmidt JH (2015) Folding of beta-barrel membrane proteins in lipid bilayers-Unassisted and assisted folding and insertion. BBA-Biomembranes 1848: 1927–1943. doi: 10.1016/j.bbamem.2015.05.004
|
| [60] |
Kleinschmidt JH (2003) Membrane protein folding on the example of outer membrane protein A of Escherichia coli. Cell Mol Life Sci 60: 1547–1558. doi: 10.1007/s00018-003-3170-0
|
| [61] |
Shahid SA, Bardiaux B, Franks WT, et al. (2012) Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods 9: 1212–1217. doi: 10.1038/nmeth.2248
|
| [62] |
Junker M, Besingi RN, Clark PL (2009) Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol Microbiol 71: 1323–1332. doi: 10.1111/j.1365-2958.2009.06607.x
|
| [63] |
Sikdar R, Peterson JH, Anderson DE, et al. (2017) Folding of a bacterial integral outer membrane protein is initiated in the periplasm. Nat Commun 8: 1309. doi: 10.1038/s41467-017-01246-4
|
| [64] | Ieva R, Skillman KM, Bernstein HD (2008) Incorporation of a polypeptide segment into the beta-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67: 188–201. |
| [65] |
Grin I, Hartmann MD, Sauer G, et al. (2014) A trimeric lipoprotein assists in trimeric autotransporter biogenesis in enterobacteria. J Biol Chem 289: 7388–7398. doi: 10.1074/jbc.M113.513275
|
| [66] |
Ishikawa M, Yoshimoto S, Hayashi A, et al. (2016) Discovery of a novel periplasmic protein that forms a complex with a trimeric autotransporter adhesin and peptidoglycan. Mol Microbiol 101: 394–410. doi: 10.1111/mmi.13398
|
| [67] |
Leo JC, Grin I, Linke D (2012) Type V secretion: mechanism(s) of autotransport through the bacterial outer membrane. Philos Trans R Soc Lond B Biol Sci 367: 1088–1101. doi: 10.1098/rstb.2011.0208
|
| [68] |
Alvarez BH, Gruber M, Ursinus A, et al. (2010) A transition from strong right-handed to canonical left-handed supercoiling in a conserved coiled-coil segment of trimeric autotransporter adhesins. J Struct Biol 170: 236–245. doi: 10.1016/j.jsb.2010.02.009
|
| [69] | Leo JC, Lyskowski A, Hattula K, et al. (2011) The structure of E. coli IgG-binding protein D suggests a general model for bending and binding in trimeric autotransporter adhesins. Structure 19: 1021–1030. |
| [70] |
Mikula KM, Leo JC, Lyskowski A, et al. (2012) The translocation domain in trimeric autotransporter adhesins is necessary and sufficient for trimerization and autotransportation. J Bacteriol 194: 827–838. doi: 10.1128/JB.05322-11
|
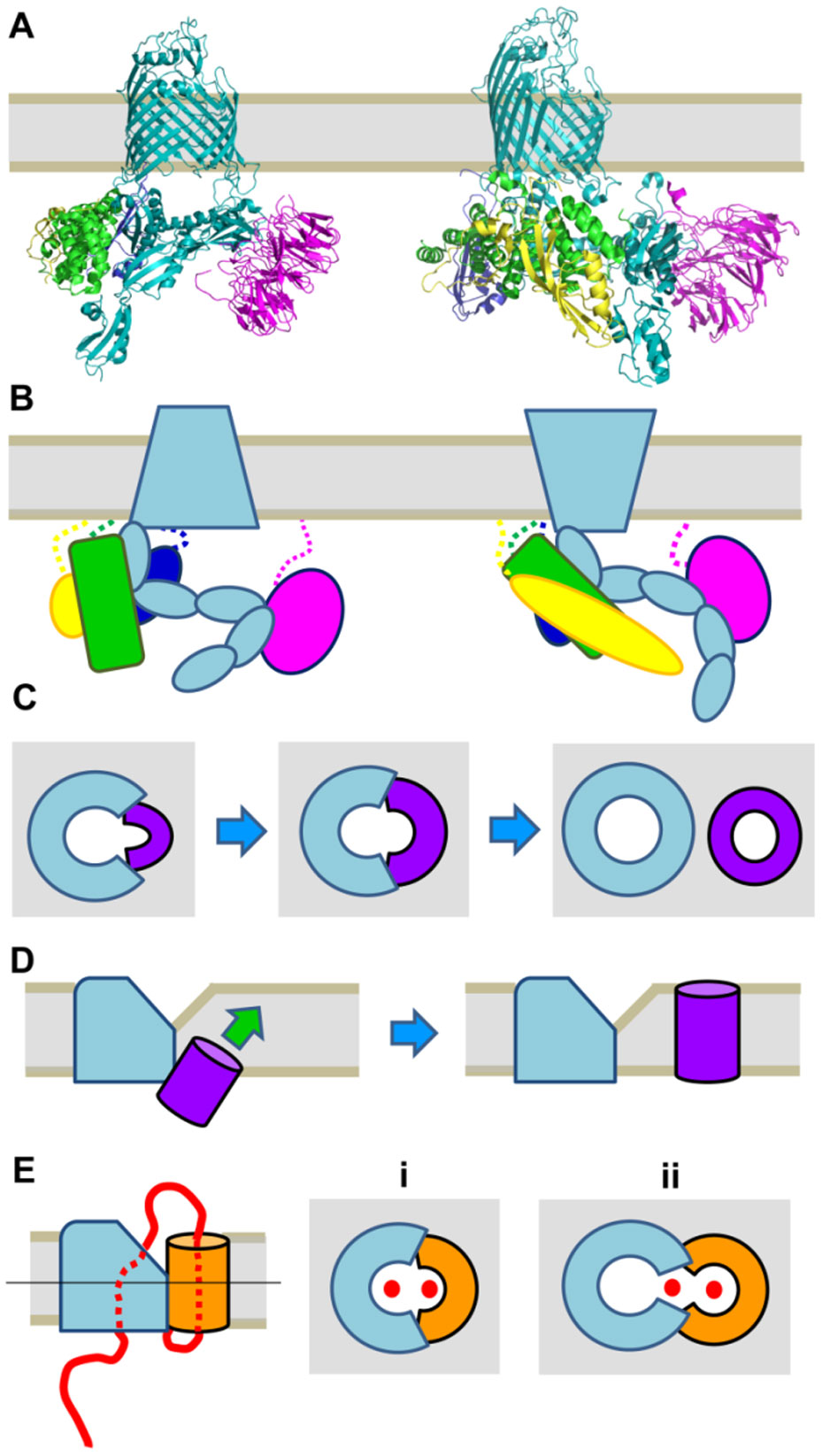
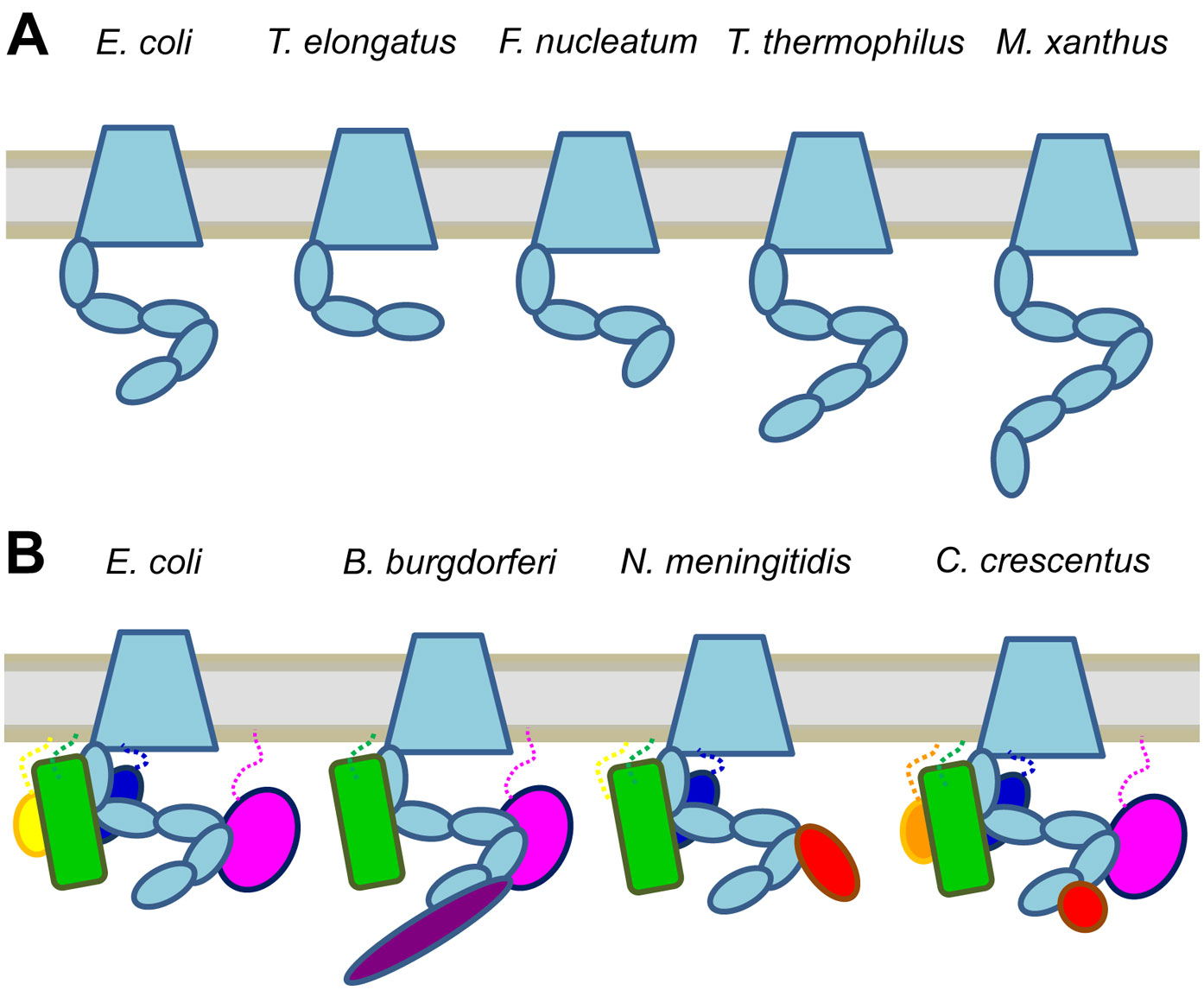
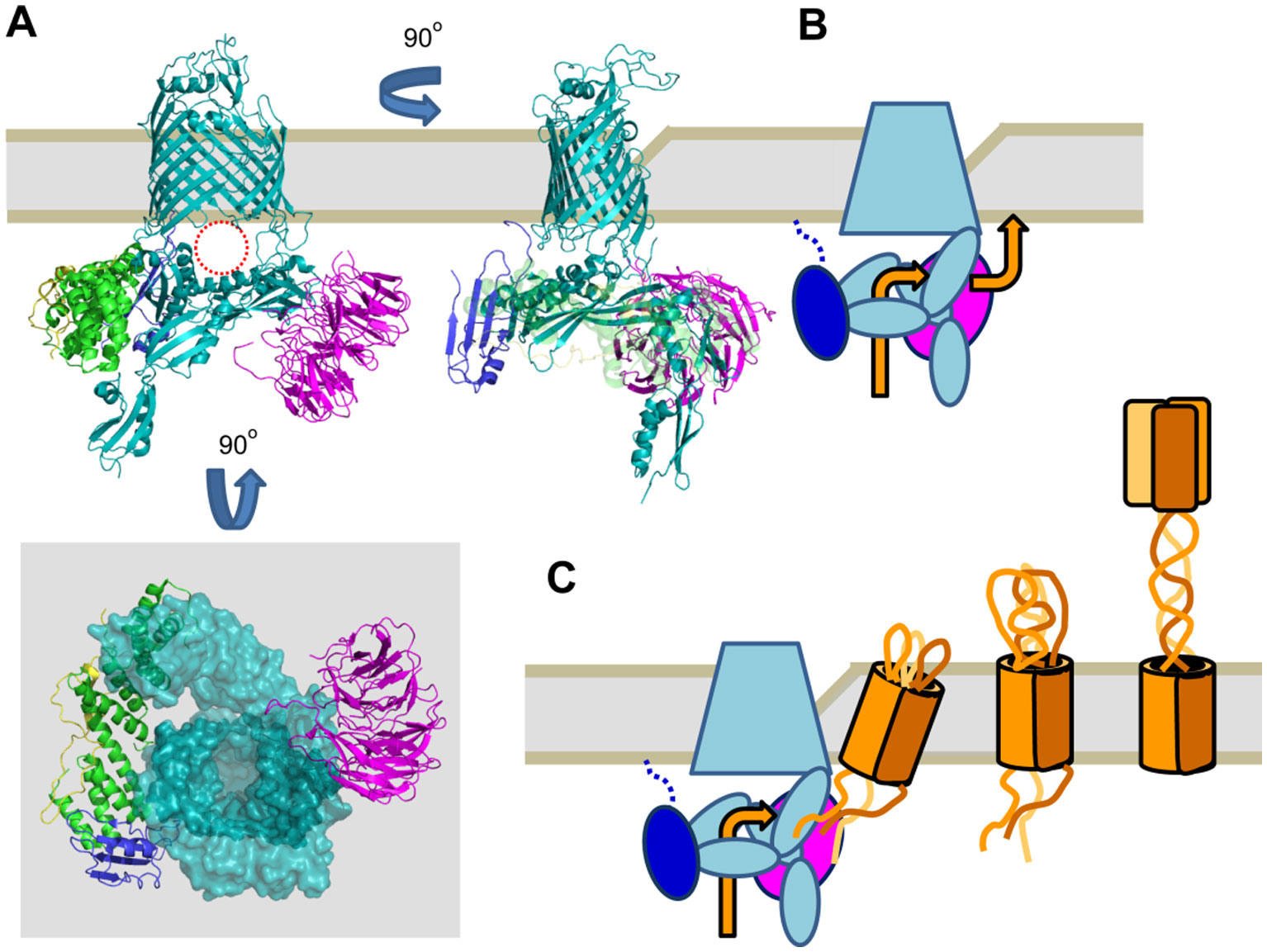
Figures(3)

Jack C. Leo, Dirk Linke. A unified model for BAM function that takes into account type Vc secretion and species differences in BAM composition[J]. AIMS Microbiology, 2018, 4(3): 455-468. doi: 10.3934/microbiol.2018.3.455







 DownLoad:
DownLoad: