Citation: Carman K.M. Ip, Jun Yin, Patrick K.S. Ng, Shiaw-Yih Lin, Gordon B. Mills. Genomic-Glycosylation Aberrations in Tumor Initiation, Progression and Management[J]. AIMS Medical Science, 2016, 3(4): 386-416. doi: 10.3934/medsci.2016.4.386

| [1] |
Gooley AA, Classon BJ, Marschalek R, et al. (1991) Glycosylation sites identified by detection of glycosylated amino acids released from Edman degradation: the identification of Xaa-Pro-Xaa-Xaa as a motif for Thr-O-glycosylation. Biochem Biophys Res Commun 178: 1194-1201. doi: 10.1016/0006-291X(91)91019-9

|
| [2] |
Yoshida A, Suzuki M, Ikenaga H, et al. (1997) Discovery of the shortest sequence motif for high level mucin-type O-glycosylation. J Biol Chem 272: 16884-16888. doi: 10.1074/jbc.272.27.16884
|
| [3] |
Young JD, Tsuchiya D, Sandlin DE, et al. (1979) Enzymic O-glycosylation of synthetic peptides from sequences in basic myelin protein. Biochemistry 18: 4444-4448. doi: 10.1021/bi00587a026
|
| [4] | Wilson IB, Gavel Y, von Heijne G (1991) Amino acid distributions around O-linked glycosylation sites. Biochem J 275 (Pt 2): 529-534. |
| [5] | Elhammer AP, Poorman RA, Brown E, et al. (1993) The specificity of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase as inferred from a database of in vivo substrates and from the in vitro glycosylation of proteins and peptides. J Biol Chem 268: 10029-10038. |
| [6] |
Elliott S, Bartley T, Delorme E, et al. (1994) Structural requirements for addition of O-linked carbohydrate to recombinant erythropoietin. Biochemistry 33: 11237-11245. doi: 10.1021/bi00203a020
|
| [7] |
O'Connell B, Tabak LA, Ramasubbu N (1991) The influence of flanking sequences on O-glycosylation. Biochem Biophys Res Commun 180: 1024-1030. doi: 10.1016/S0006-291X(05)81168-4
|
| [8] |
Lehle L BE (1984) Primary structural requirements for N- and O-glycosylation of yeast mannoproteins. Biochim Biophys Acta 799: 246-251. doi: 10.1016/0304-4165(84)90267-8
|
| [9] |
Ladenson RP, Schwartz SO, Ivy AC (1949) Incidence of the blood groups and the secretor factor in patients with pernicious anemia and stomach carcinoma. Am J Med Sci 217: 194-197. doi: 10.1097/00000441-194902000-00011
|
| [10] |
Hakomori SI, Murakami WT (1968) Glycolipids of hamster fibroblasts and derived malignant-transformed cell lines. Proc Natl Acad Sci U S A 59: 254-261. doi: 10.1073/pnas.59.1.254
|
| [11] |
Hakomori S (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A 99: 10231-10233. doi: 10.1073/pnas.172380699
|
| [12] |
Reis CA, Osorio H, Silva L, et al. (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 63: 322-329. doi: 10.1136/jcp.2009.071035
|
| [13] |
Ma Z, Vosseller K (2014) Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 289: 34457-34465. doi: 10.1074/jbc.R114.577718
|
| [14] |
Fuster MM, Esko JD (2005) The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer 5: 526-542. doi: 10.1038/nrc1649
|
| [15] |
Kandoth C, McLellan MD, Vandin F, et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502: 333-339. doi: 10.1038/nature12634
|
| [16] |
Anwar A, Norris DA, Fujita M (2011) Ubiquitin proteasomal pathway mediated degradation of p53 in melanoma. Arch Biochem Biophys 508: 198-203. doi: 10.1016/j.abb.2010.12.012
|
| [17] |
Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594-604. doi: 10.1038/nrc864
|
| [18] |
Riley T, Sontag E, Chen P, et al. (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402-412. doi: 10.1038/nrm2395
|
| [19] |
Vousden KH, Prives C (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137: 413-431. doi: 10.1016/j.cell.2009.04.037
|
| [20] |
Yang WH, Kim JE, Nam HW, et al. (2006) Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol 8: 1074-1083. doi: 10.1038/ncb1470
|
| [21] | Shaw P, Freeman J, Bovey R, et al. (1996) Regulation of specific DNA binding by p53: evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene 12: 921-930. |
| [22] |
Ito A, Lai CH, Zhao X, et al. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20: 1331-1340. doi: 10.1093/emboj/20.6.1331
|
| [23] |
Rodriguez MS, Desterro JM, Lain S, et al. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol 20: 8458-8467. doi: 10.1128/MCB.20.22.8458-8467.2000
|
| [24] |
Chuikov S, Kurash JK, Wilson JR, et al. (2004) Regulation of p53 activity through lysine methylation. Nature 432: 353-360. doi: 10.1038/nature03117
|
| [25] |
Haupt Y, Maya R, Kazaz A, et al. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296-299. doi: 10.1038/387296a0
|
| [26] |
Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299-303. doi: 10.1038/387299a0
|
| [27] |
Bode AM, Dong Z (2004) Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4: 793-805. doi: 10.1038/nrc1455
|
| [28] |
Bech-Otschir D, Kraft R, Huang X, et al. (2001) COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J 20: 1630-1639. doi: 10.1093/emboj/20.7.1630
|
| [29] |
Liu K, Paterson AJ, Zhang F, et al. (2004) Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J Neurochem 89: 1044-1055. doi: 10.1111/j.1471-4159.2004.02389.x
|
| [30] | Muller PA, Vousden KH (2013) p53 mutations in cancer. Nat Cell Biol 15: 2-8. |
| [31] |
Gaiddon C, Lokshin M, Ahn J, et al. (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21: 1874-1887. doi: 10.1128/MCB.21.5.1874-1887.2001
|
| [32] |
Strano S, Munarriz E, Rossi M, et al. (2000) Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 275: 29503-29512. doi: 10.1074/jbc.M003360200
|
| [33] |
Bian YS, Osterheld MC, Bosman FT, et al. (2001) p53 gene mutation and protein accumulation during neoplastic progression in Barrett's esophagus. Mod Pathol 14: 397-403. doi: 10.1038/modpathol.3880324
|
| [34] |
Maehama T, Dixon JE. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375-13378. doi: 10.1074/jbc.273.22.13375
|
| [35] |
Maehama T, Taylor GS, Dixon JE (2001) PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem 70: 247-279. doi: 10.1146/annurev.biochem.70.1.247
|
| [36] | Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606-619. |
| [37] |
Trotman LC, Wang X, Alimonti A, et al. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128: 141-156. doi: 10.1016/j.cell.2006.11.040
|
| [38] |
Gong L, Govan JM, Evans EB, et al. (2015) Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle 14: 2323-2332. doi: 10.1080/15384101.2015.1044174
|
| [39] |
Shi Y, Paluch BE, Wang X, et al. (2012) PTEN at a glance. J Cell Sci 125: 4687-4692. doi: 10.1242/jcs.093765
|
| [40] | Bermudez Brito M, Goulielmaki E, Papakonstanti EA (2015) Focus on PTEN Regulation. Front Oncol 5: 166. |
| [41] |
Hopkins BD, Fine B, Steinbach N, et al. (2013) A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341: 399-402. doi: 10.1126/science.1234907
|
| [42] |
Wang X, Trotman LC, Koppie T, et al. (2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128: 129-139. doi: 10.1016/j.cell.2006.11.039
|
| [43] |
Vazquez F, Ramaswamy S, Nakamura N, et al. (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010-5018. doi: 10.1128/MCB.20.14.5010-5018.2000
|
| [44] | Torres J, Pulido R (2001) The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 276: 993-998. |
| [45] | Tolkacheva T, Boddapati M, Sanfiz A, et al. (2001) Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res 61: 4985-4989. |
| [46] |
Vazquez F, Grossman SR, Takahashi Y, et al. (2001) Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 276: 48627-48630. doi: 10.1074/jbc.C100556200
|
| [47] |
Das S, Dixon JE, Cho W (2003) Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A 100: 7491-7496. doi: 10.1073/pnas.0932835100
|
| [48] |
Vazquez F, Matsuoka S, Sellers WR, et al. (2006) Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci U S A 103: 3633-3638. doi: 10.1073/pnas.0510570103
|
| [49] |
Maccario H, Perera NM, Davidson L, et al. (2007) PTEN is destabilized by phosphorylation on Thr366. Biochem J 405: 439-444. doi: 10.1042/BJ20061837
|
| [50] |
Xu D, Yao Y, Jiang X, et al. (2010) Regulation of PTEN stability and activity by Plk3. J Biol Chem 285: 39935-39942. doi: 10.1074/jbc.M110.166462
|
| [51] |
Gil A, Andres-Pons A, Fernandez E, et al. (2006) Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell 17: 4002-4013. doi: 10.1091/mbc.E06-05-0380
|
| [52] |
Ding L, Chen S, Liu P, et al. (2014) CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res 74: 2050-2061. doi: 10.1158/0008-5472.CAN-13-1659
|
| [53] |
Wang Z, Pandey A, Hart GW (2007) Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol Cell Proteomics 6: 1365-1379. doi: 10.1074/mcp.M600453-MCP200
|
| [54] |
Zeidan Q, Hart GW (2010) The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci 123: 13-22. doi: 10.1242/jcs.053678
|
| [55] |
Wang Z, Gucek M, Hart GW (2008) Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A 105: 13793-13798. doi: 10.1073/pnas.0806216105
|
| [56] |
Waite KA, Eng C (2002) Protean PTEN: form and function. Am J Hum Genet 70: 829-844. doi: 10.1086/340026
|
| [57] | Han SY, Kato H, Kato S, et al. (2000) Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res 60: 3147-3151. |
| [58] |
Georgescu MM, Kirsch KH, Akagi T, et al. (1999) The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A 96: 10182-10187. doi: 10.1073/pnas.96.18.10182
|
| [59] | Zhao Y, Huang Q, Wang A, et al. (2009) Mutation of PTEN in glioma stem/progenitor cells: a case report. Cancer Genet Cytogenet 195: 183-185. |
| [60] | Fan X, Munoz J, Sanko SG, et al. (2002) PTEN, DMBT1, and p16 alterations in diffusely infiltrating astrocytomas. Int J Oncol 21: 667-674. |
| [61] | Trigka EA, Levidou G, Saetta AA, et al. (2013) A detailed immunohistochemical analysis of the PI3K/AKT/mTOR pathway in lung cancer: correlation with PIK3CA, AKT1, K-RAS or PTEN mutational status and clinicopathological features. Oncol Rep 30: 623-636. |
| [62] |
Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061-1068. doi: 10.1038/nature07385
|
| [63] |
Cancer Genome Atlas Research N (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609-615. doi: 10.1038/nature10166
|
| [64] |
Wallace MD, Pfefferle AD, Shen L, et al. (2012) Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics 192: 385-396. doi: 10.1534/genetics.112.142802
|
| [65] |
Ding L, Getz G, Wheeler DA, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069-1075. doi: 10.1038/nature07423
|
| [66] |
Boudry-Labis E, Roche-Lestienne C, Nibourel O, et al. (2013) Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am J Hematol 88: 306-311. doi: 10.1002/ajh.23403
|
| [67] |
Martin GA, Viskochil D, Bollag G, et al. (1990) The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63: 843-849. doi: 10.1016/0092-8674(90)90150-D
|
| [68] | Xu GF, Lin B, Tanaka K, et al. (1990) The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63: 835-841. |
| [69] |
Izawa I, Tamaki N, Saya H (1996) Phosphorylation of neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett 382: 53-59. doi: 10.1016/0014-5793(96)00137-8
|
| [70] |
Feng L, Yunoue S, Tokuo H, et al. (2004) PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett 557: 275-282. doi: 10.1016/S0014-5793(03)01507-2
|
| [71] |
Nasir ud D, Kaleem A, Ahmad I, et al. (2009) Effect on the Ras/Raf signaling pathway of post-translational modifications of neurofibromin: in silico study of protein modification responsible for regulatory pathways. J Cell Biochem 108: 816-824. doi: 10.1002/jcb.22301
|
| [72] |
Griffith LS, Schmitz B. (1999) O-linked N-acetylglucosamine levels in cerebellar neurons respond reciprocally to pertubations of phosphorylation. Eur J Biochem 262: 824-831. doi: 10.1046/j.1432-1327.1999.00439.x
|
| [73] |
Mangoura D, Sun Y, Li C, et al. (2006) Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 25: 735-745. doi: 10.1038/sj.onc.1209113
|
| [74] |
Leondaritis G, Petrikkos L, Mangoura D (2009) Regulation of the Ras-GTPase activating protein neurofibromin by C-tail phosphorylation: implications for protein kinase C/Ras/extracellular signal-regulated kinase 1/2 pathway signaling and neuronal differentiation. J Neurochem 109: 573-583. doi: 10.1111/j.1471-4159.2009.05975.x
|
| [75] | Kaufmann D, Junge I, Bartelt B, et al. (1999) On the lysosomal degradation of neurofibromin and its phosphorylation in cultured melanocytes. Biol Chem 380: 1071-1078. |
| [76] |
Perlman SL, Boder Deceased E, Sedgewick RP, et al. (2012) Ataxia-telangiectasia. Handb Clin Neurol 103: 307-332. doi: 10.1016/B978-0-444-51892-7.00019-X
|
| [77] |
McKinnon PJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7: 303-321. doi: 10.1146/annurev-pathol-011811-132509
|
| [78] |
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology 14: 197-210. doi: 10.1038/nrm3546
|
| [79] |
Ahmed M, Rahman N (2006) ATM and breast cancer susceptibility. Oncogene 25: 5906-5911. doi: 10.1038/sj.onc.1209873
|
| [80] | Swift M, Morrell D, Cromartie E, et al. (1986) The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet 39: 573-583. |
| [81] |
Renwick A, Thompson D, Seal S, et al. (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38: 873-875. doi: 10.1038/ng1837
|
| [82] | Morrell D, Cromartie E, Swift M (1986) Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst 77: 89-92. |
| [83] |
Abraham RT (2004) PI 3-kinase related kinases: 'big' players in stress-induced signaling pathways. DNA Repair (Amst) 3: 883-887. doi: 10.1016/j.dnarep.2004.04.002
|
| [84] |
Paull TT (2015) Mechanisms of ATM Activation. Annu Rev Biochem 84: 711-738. doi: 10.1146/annurev-biochem-060614-034335
|
| [85] | Jung M, Kondratyev A, Lee SA, et al. (1997) ATM gene product phosphorylates I kappa B-alpha. Cancer Res 57: 24-27. |
| [86] |
Li S, Ting NS, Zheng L, et al. (2000) Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 406: 210-215. doi: 10.1038/35018134
|
| [87] |
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499-506. doi: 10.1038/nature01368
|
| [88] |
Sun Y, Xu Y, Roy K, et al. (2007) DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 27: 8502-8509. doi: 10.1128/MCB.01382-07
|
| [89] |
Kozlov SV, Graham ME, Jakob B, et al. (2011) Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem 286: 9107-9119. doi: 10.1074/jbc.M110.204065
|
| [90] |
Miura Y, Sakurai Y, Endo T (2012) O-GlcNAc modification affects the ATM-mediated DNA damage response. Biochim Biophys Acta 1820: 1678-1685. doi: 10.1016/j.bbagen.2012.06.013
|
| [91] |
Zhong J, Martinez M, Sengupta S, et al. (2015) Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics 15: 591-607. doi: 10.1002/pmic.201400339
|
| [92] | Easton DF, Ford D, Bishop DT (1995) Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet 56: 265-271. |
| [93] | Schubert EL, Lee MK, Mefford HC, et al. (1997) BRCA2 in American families with four or more cases of breast or ovarian cancer: recurrent and novel mutations, variable expression, penetrance, and the possibility of families whose cancer is not attributable to BRCA1 or BRCA2. Am J Hum Genet 60: 1031-1040. |
| [94] |
Hennessy BT, Timms KM, Carey MS, et al. (2010) Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clinic Oncology offic J Am Soc Clin Oncology 28: 3570-3576. doi: 10.1200/JCO.2009.27.2997
|
| [95] |
Gonzalez-Angulo AM, Timms KM, Liu S, et al. (2011) Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res 17: 1082-1089. doi: 10.1158/1078-0432.CCR-10-2560
|
| [96] |
Huen MS, Sy SM, Chen J (2010) BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Molecular Cell Biology 11: 138-148. doi: 10.1038/nrm2831
|
| [97] |
Rahman N, Stratton MR (1998) The genetics of breast cancer susceptibility. Annu Rev Genet 32: 95-121. doi: 10.1146/annurev.genet.32.1.95
|
| [98] |
Kleiman FE, Manley JL (2001) The BARD1-CstF-50 interaction links mRNA 3' end formation to DNA damage and tumor suppression. Cell 104: 743-753. doi: 10.1016/S0092-8674(01)00270-7
|
| [99] |
Welcsh PL, King MC (2001) BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 10: 705-713. doi: 10.1093/hmg/10.7.705
|
| [100] |
Chen PL, Chen CF, Chen Y, et al. (1998) The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci U S A 95: 5287-5292. doi: 10.1073/pnas.95.9.5287
|
| [101] |
Savage KI, Harkin DP (2015) BRCA1, a 'complex' protein involved in the maintenance of genomic stability. FEBS J 282: 630-646. doi: 10.1111/febs.13150
|
| [102] |
Tram E, Savas S, Ozcelik H (2013) Missense variants of uncertain significance (VUS) altering the phosphorylation patterns of BRCA1 and BRCA2. PLoS One 8: e62468. doi: 10.1371/journal.pone.0062468
|
| [103] | Jensen RA TM, Jetton TL, Szabo CI, et al. (1996) BRCA1 is secreted and exhibits properties of a granin. Nature 12: 303-308. |
| [104] |
Whelan SA, Lu M, He J, et al. (2009) Mass spectrometry (LC-MS/MS) site-mapping of N-glycosylated membrane proteins for breast cancer biomarkers. J Proteome Res 8: 4151-4160. doi: 10.1021/pr900322g
|
| [105] |
Scully R, Chen J, Plug A, et al. (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265-275. doi: 10.1016/S0092-8674(00)81847-4
|
| [106] | Siddique H, Rao VN, Reddy ES (2009) CBP-mediated post-translational N-glycosylation of BRCA2. Int J Oncol 35: 387-391. |
| [107] |
Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2: 103-112. doi: 10.1016/S1535-6108(02)00102-2
|
| [108] |
Di Fiore R, D'Anneo A, Tesoriere G, et al. (2013) RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol 228: 1676-1687. doi: 10.1002/jcp.24329
|
| [109] |
Liu H, Dibling B, Spike B, et al. (2004) New roles for the RB tumor suppressor protein. Curr Opin Genet Dev 14: 55-64. doi: 10.1016/j.gde.2003.11.005
|
| [110] |
Giacinti C, Giordano A (2006) RB and cell cycle progression. Oncogene 25: 5220-5227. doi: 10.1038/sj.onc.1209615
|
| [111] |
Dowdy SF, Hinds PW, Louie K, et al. (1993) Physical interaction of the retinoblastoma protein with human D cyclins. Cell 73: 499-511. doi: 10.1016/0092-8674(93)90137-F
|
| [112] |
De Luca A, MacLachlan TK, Bagella L, et al. (1997) A unique domain of pRb2/p130 acts as an inhibitor of Cdk2 kinase activity. J Biol Chem 272: 20971-20974. doi: 10.1074/jbc.272.34.20971
|
| [113] | Chan HM, La Thangue NB (2001) p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363-2373. |
| [114] |
Carr SM, Munro S, Kessler B, et al. (2011) Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J 30: 317-327. doi: 10.1038/emboj.2010.311
|
| [115] |
Ledl A, Schmidt D, Muller S (2005) Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene 24: 3810-3818. doi: 10.1038/sj.onc.1208539
|
| [116] |
Wells L, Slawson C, Hart GW (2011) The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids 40: 877-883. doi: 10.1007/s00726-010-0709-x
|
| [117] |
Delston RB, Matatall KA, Sun Y, et al. (2011) p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene 30: 588-599. doi: 10.1038/onc.2010.442
|
| [118] |
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127-137. doi: 10.1038/35052073
|
| [119] |
Contessa JN, Bhojani MS, Freeze HH, et al. (2008) Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res 68: 3803-3809. doi: 10.1158/0008-5472.CAN-07-6389
|
| [120] | Soderquist AM, Carpenter G (1984) Glycosylation of the epidermal growth factor receptor in A-431 cells. The contribution of carbohydrate to receptor function. J Biol Chem 259: 12586-12594. |
| [121] | Slieker LJ, Martensen TM, Lane MD (1986) Synthesis of epidermal growth factor receptor in human A431 cells. Glycosylation-dependent acquisition of ligand binding activity occurs post-translationally in the endoplasmic reticulum. J Biol Chem 261: 15233-15241. |
| [122] |
Yokoe S, Takahashi M, Asahi M, et al. (2007) The Asn418-linked N-glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res 67: 1935-1942. doi: 10.1158/0008-5472.CAN-06-3023
|
| [123] | Tsuda T, Ikeda Y, Taniguchi N (2000) The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J Biol Chem 275: 21988-21994. |
| [124] |
Fernandes H, Cohen S, Bishayee S (2001) Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem 276: 5375-5383. doi: 10.1074/jbc.M005599200
|
| [125] | Ekstrand AJ, Liu L, He J, et al. (1995) Altered subcellular location of an activated and tumour-associated epidermal growth factor receptor. Oncogene 10: 1455-1460. |
| [126] | Moscatello DK, Montgomery RB, Sundareshan P, et al. (1996) Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene 13: 85-96. |
| [127] |
Nishikawa R, Ji XD, Harmon RC, et al. (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A 91: 7727-7731. doi: 10.1073/pnas.91.16.7727
|
| [128] |
Humphrey PA, Gangarosa LM, Wong AJ, et al. (1991) Deletion-mutant epidermal growth factor receptor in human gliomas: effects of type II mutation on receptor function. Biochem Biophys Res Commun 178: 1413-1420. doi: 10.1016/0006-291X(91)91051-D
|
| [129] |
Saxon ML, Lee DC (1999) Mutagenesis reveals a role for epidermal growth factor receptor extracellular subdomain IV in ligand binding. J Biol Chem 274: 28356-28362. doi: 10.1074/jbc.274.40.28356
|
| [130] |
Domagala T, Konstantopoulos N, Smyth F, et al. (2000) Stoichiometry, kinetic and binding analysis of the interaction between epidermal growth factor (EGF) and the extracellular domain of the EGF receptor. Growth factors 18: 11-29. doi: 10.3109/08977190009003231
|
| [131] |
Elleman TC, Domagala T, McKern NM, et al. (2001) Identification of a determinant of epidermal growth factor receptor ligand-binding specificity using a truncated, high-affinity form of the ectodomain. Biochemistry 40: 8930-8939. doi: 10.1021/bi010037b
|
| [132] |
Whitson KB, Whitson SR, Red-Brewer ML, et al. (2005) Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor. Biochemistry 44: 14920-14931. doi: 10.1021/bi050751j
|
| [133] |
Yoon SJ, Nakayama K, Hikita T, et al. (2006) Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc Natl Acad Sci U S A 103: 18987-18991. doi: 10.1073/pnas.0609281103
|
| [134] |
Wakshull EM, Wharton W (1985) Stabilized complexes of epidermal growth factor and its receptor on the cell surface stimulate RNA synthesis but not mitogenesis. Proc Natl Acad Sci U S A 82: 8513-8517. doi: 10.1073/pnas.82.24.8513
|
| [135] |
Hirabayashi J, Hashidate T, Arata Y, et al. (2002) Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta 1572: 232-254. doi: 10.1016/S0304-4165(02)00311-2
|
| [136] |
Partridge EA, Le Roy C, Di Guglielmo GM, et al. (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306: 120-124. doi: 10.1126/science.1102109
|
| [137] |
Lajoie P, Partridge EA, Guay G, et al. (2007) Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol 179: 341-356. doi: 10.1083/jcb.200611106
|
| [138] |
Guo HB, Johnson H, Randolph M, et al. (2009) Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 19: 547-559. doi: 10.1093/glycob/cwp023
|
| [139] |
Guo HB, Randolph M, Pierce M (2007) Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J Biol Chem 282: 22150-22162. doi: 10.1074/jbc.M611518200
|
| [140] | Guo P, Wang QY, Guo HB, et al. (2004) N-acetylglucosaminyltransferase V modifies the signaling pathway of epidermal growth factor receptor. Cell Mol Life Sci 61: 1795-1804. |
| [141] |
Guo H, Nairn A, dela Rosa M, et al. (2012) Transcriptional regulation of the protocadherin beta cluster during Her-2 protein-induced mammary tumorigenesis results from altered N-glycan branching. J Biol Chem 287: 24941-24954. doi: 10.1074/jbc.M112.369355
|
| [142] |
Guo HB, Johnson H, Randolph M, et al. (2010) Specific posttranslational modification regulates early events in mammary carcinoma formation. Proc Natl Acad Sci U S A 107: 21116-21121. doi: 10.1073/pnas.1013405107
|
| [143] |
Liu YC, Yen HY, Chen CY, et al. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A 108: 11332-11337. doi: 10.1073/pnas.1107385108
|
| [144] |
Yen HY, Liu YC, Chen NY, et al. (2015) Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc Natl Acad Sci U S A 112: 6955-6960. doi: 10.1073/pnas.1507329112
|
| [145] |
Matsumoto K, Yokote H, Arao T, et al. (2008) N-Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci 99: 1611-1617. doi: 10.1111/j.1349-7006.2008.00847.x
|
| [146] |
Lin WL, Lin YS, Shi GY, et al. (2015) Lewisy promotes migration of oral cancer cells by glycosylation of epidermal growth factor receptor. PLoS One 10: e0120162. doi: 10.1371/journal.pone.0120162
|
| [147] |
Ling YH, Li T, Perez-Soler R, et al. (2009) Activation of ER stress and inhibition of EGFR N-glycosylation by tunicamycin enhances susceptibility of human non-small cell lung cancer cells to erlotinib. Cancer Chemotherapy Pharmacol 64: 539-548. doi: 10.1007/s00280-008-0902-8
|
| [148] |
Park JJ, Yi JY, Jin YB, et al. (2012) Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem Pharmacol 83: 849-857. doi: 10.1016/j.bcp.2012.01.007
|
| [149] | Gan HK, Walker F, Burgess AW, et al. (2007) The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J Biol Chem 282: 2840-2850. |
| [150] |
Sprung R, Nandi A, Chen Y, et al. (2005) Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J Proteome Res 4: 950-957. doi: 10.1021/pr050033j
|
| [151] |
Stateva SR, Villalobo A (2015) O-GlcNAcylation of the human epidermal growth factor receptor. Organ Biomol Chem 13: 8196-8204. doi: 10.1039/C5OB00443H
|
| [152] | Kaleem A, Ahmad I, Hoessli DC, et al. (2009) Epidermal growth factor receptors: function modulation by phosphorylation and glycosylation interplay. Mol Biol Rep 36: 631-639. |
| [153] |
Bao J, Alroy I, Waterman H, et al. (2000) Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J Biol Chem 275: 26178-26186. doi: 10.1074/jbc.M002367200
|
| [154] |
Feinmesser RL, Wicks SJ, Taverner CJ, et al. (1999) Ca2+/calmodulin-dependent kinase II phosphorylates the epidermal growth factor receptor on multiple sites in the cytoplasmic tail and serine 744 within the kinase domain to regulate signal generation. J Biol Chem 274: 16168-16173. doi: 10.1074/jbc.274.23.16168
|
| [155] | Countaway JL, Nairn AC, Davis RJ (1992) Mechanism of desensitization of the epidermal growth factor receptor protein-tyrosine kinase. J Biol Chem 267: 1129-1140. |
| [156] |
Jaiswal BS, Kljavin NM, Stawiski EW, et al. (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23: 603-617. doi: 10.1016/j.ccr.2013.04.012
|
| [157] |
Prickett TD, Agrawal NS, Wei X, et al. (2009) Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet 41: 1127-1132. doi: 10.1038/ng.438
|
| [158] |
Kurppa KJ, Denessiouk K, Johnson MS, et al. (2016) Activating ERBB4 mutations in non-small cell lung cancer. Oncogene 35: 1283-1291. doi: 10.1038/onc.2015.185
|
| [159] | Polakis P (2012) Wnt signaling in cancer. Cold Spring Harbor perspectives in biology 4. |
| [160] |
Liu C, Li Y, Semenov M, et al. (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108: 837-847. doi: 10.1016/S0092-8674(02)00685-2
|
| [161] |
Winer IS, Bommer GT, Gonik N, et al. (2006) Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J Biol Chem 281: 26181-26187. doi: 10.1074/jbc.M604217200
|
| [162] |
Li VS, Ng SS, Boersema PJ, et al. (2012) Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 149: 1245-1256. doi: 10.1016/j.cell.2012.05.002
|
| [163] |
Gao C, Xiao G, Hu J (2014) Regulation of Wnt/beta-catenin signaling by posttranslational modifications. Cell Biosci 4: 13. doi: 10.1186/2045-3701-4-13
|
| [164] |
Zhu W, Leber B, Andrews DW (2001) Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J 20: 5999-6007. doi: 10.1093/emboj/20.21.5999
|
| [165] |
Sayat R, Leber B, Grubac V, et al. (2008) O-GlcNAc-glycosylation of beta-catenin regulates its nuclear localization and transcriptional activity. Experiment Cell Res 314: 2774-2787. doi: 10.1016/j.yexcr.2008.05.017
|
| [166] |
Olivier-Van Stichelen S, Dehennaut V, Buzy A, et al. (2014) O-GlcNAcylation stabilizes beta-catenin through direct competition with phosphorylation at threonine 41. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 28: 3325-3338. doi: 10.1096/fj.13-243535
|
| [167] |
Vosseller K, Wells L, Lane MD, et al. (2002) Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A 99: 5313-5318. doi: 10.1073/pnas.072072399
|
| [168] |
Ha JR, Hao L, Venkateswaran G, et al. (2014) beta-catenin is O-GlcNAc glycosylated at Serine 23: implications for beta-catenin's subcellular localization and transactivator function. Experiment Cell Res 321: 153-166. doi: 10.1016/j.yexcr.2013.11.021
|
| [169] |
van Noort M, van de Wetering M, Clevers H (2002) Identification of two novel regulated serines in the N terminus of beta-catenin. Experiment Cell Res 276: 264-272. doi: 10.1006/excr.2002.5520
|
| [170] | Nhieu JT, Renard CA, Wei Y, et al. (1999) Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol 155: 703-710. |
| [171] |
Mazumder R, Morampudi KS, Motwani M, et al. (2012) Proteome-wide analysis of single-nucleotide variations in the N-glycosylation sequon of human genes. PLoS One 7: e36212. doi: 10.1371/journal.pone.0036212
|
| [172] |
Karagiannis K, Simonyan V, Mazumder R (2013) SNVDis: a proteome-wide analysis service for evaluating nsSNVs in protein functional sites and pathways. Genom Proteom Bioinform 11: 122-126. doi: 10.1016/j.gpb.2012.10.003
|
| [173] |
Cole C, Krampis K, Karagiannis K, et al. (2014) Non-synonymous variations in cancer and their effects on the human proteome: workflow for NGS data biocuration and proteome-wide analysis of TCGA data. BMC Bioinform 15: 28. doi: 10.1186/1471-2105-15-28
|
| [174] | Wu TJ, Shamsaddini A, Pan Y, et al. (2014) A framework for organizing cancer-related variations from existing databases, publications and NGS data using a High-performance Integrated Virtual Environment (HIVE). Database J Biolog Database Curation 2014: bau022. |
| [175] |
Meany DL, Chan DW (2011) Aberrant glycosylation associated with enzymes as cancer biomarkers. Clinical Proteom 8: 7. doi: 10.1186/1559-0275-8-7
|
| [176] | Venkitachalam S, Revoredo L, Varadan V, et al. (2016) Biochemical and functional characterization of glycosylation-associated mutational landscapes in colon cancer. Sci Report 6: 23642. |
| [177] |
Tanizaki J, Ercan D, Capelletti M, et al. (2015) Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res 75: 3139-3146. doi: 10.1158/0008-5472.CAN-14-3771
|
| [178] |
Yang X, Ongusaha PP, Miles PD, et al. (2008) Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451: 964-969. doi: 10.1038/nature06668
|
| [179] |
Federici M, Menghini R, Mauriello A, et al. (2002) Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 106: 466-472. doi: 10.1161/01.CIR.0000023043.02648.51
|
| [180] |
Wang S, Huang X, Sun D, et al. (2012) Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS One 7: e37427. doi: 10.1371/journal.pone.0037427
|
| [181] |
Licitra L, Suardi S, Bossi P, et al. (2004) Prediction of TP53 status for primary cisplatin, fluorouracil, and leucovorin chemotherapy in ethmoid sinus intestinal-type adenocarcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 22: 4901-4906. doi: 10.1200/JCO.2004.05.071
|
| [182] | Munirajan AK, Tutsumi-Ishii Y, Mohanprasad BK, et al. (1996) p53 gene mutations in oral carcinomas from India. Int J Cancer 66: 297-300. |
| [183] | Balz V, Scheckenbach K, Gotte K, et al. (2003) Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2-11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res 63: 1188-1191. |
| [184] |
[Hsu CH, Yang SA, Wang JY, et al. (1999) Mutational spectrum of p53 gene in arsenic-related skin cancers from the blackfoot disease endemic area of Taiwan. Br J Cancer 80: 1080-1086. doi: 10.1038/sj.bjc.6690467
|
| [185] |
Saito T, Oda Y, Kawaguchi K, et al. (2003) PTEN/MMAC1 gene mutation is a rare event in soft tissue sarcomas without specific balanced translocations. Int J Cancer 104: 175-178. doi: 10.1002/ijc.10918
|
| [186] |
Crona J, Verdugo AD, Granberg D, et al. (2013) Next-generation sequencing in the clinical genetic screening of patients with pheochromocytoma and paraganglioma. Endocr Connect 2: 104-111. doi: 10.1530/EC-13-0009
|
| [187] |
Beltrame L, Di Marino M, Fruscio R, et al. (2015) Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: a retrospective study. Ann Oncol 26: 1363-1371. doi: 10.1093/annonc/mdv472.137
|
| [188] |
Ciriello G, Gatza ML, Beck AH, et al. (2015) Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163: 506-519. doi: 10.1016/j.cell.2015.09.033
|
| [189] |
Papaemmanuil E, Gerstung M, Malcovati L, et al. (2013) Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122: 3616-3627; quiz 3699. doi: 10.1182/blood-2013-08-518886
|
| [190] |
Cancer Genome Atlas Research N, Kandoth C, Schultz N, et al. (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497: 67-73. doi: 10.1038/nature12113
|
| [191] |
Frattini V, Trifonov V, Chan JM, et al. (2013) The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45: 1141-1149. doi: 10.1038/ng.2734
|
| [192] |
George J, Lim JS, Jang SJ, et al. (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524: 47-53. doi: 10.1038/nature14664
|
| [193] |
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507: 315-322. doi: 10.1038/nature12965
|
| [194] |
Dulak AM, Stojanov P, Peng S, et al. (2013) Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 45: 478-486. doi: 10.1038/ng.2591
|
| [195] |
Cancer Genome Atlas N (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330-337. doi: 10.1038/nature11252
|
| [196] |
Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61-70. doi: 10.1038/nature11412
|
| [197] |
Pickering CR, Zhou JH, Lee JJ, et al. (2014) Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res 20: 6582-6592. doi: 10.1158/1078-0432.CCR-14-1768
|
| [198] |
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513: 202-209. doi: 10.1038/nature13480
|
| [199] | Giannakis M, Mu XJ, Shukla SA, et al. (2016) Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. |
| [200] | Quesada V, Conde L, Villamor N, et al. (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47-52. |
| [201] |
Zang ZJ, Cutcutache I, Poon SL, et al. (2012) Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet 44: 570-574. doi: 10.1038/ng.2246
|
| [202] |
Li YY, Hanna GJ, Laga AC, et al. (2015) Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res 21: 1447-1456. doi: 10.1158/1078-0432.CCR-14-1773
|
| [203] | Haruta M, Arai Y, Watanabe N, et al. (2012) Different incidences of epigenetic but not genetic abnormalities between Wilms tumors in Japanese and Caucasian children. Cancer Sci 103: 1129-1135. |
| [204] |
Laurent-Puig P, Legoix P, Bluteau O, et al. (2001) Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 120: 1763-1773. doi: 10.1053/gast.2001.24798
|
| [205] |
Boyault S, Rickman DS, de Reynies A, et al. (2007) Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45: 42-52. doi: 10.1002/hep.21467
|
| [206] |
Kazakov DV, Sima R, Vanecek T, et al. (2009) Mutations in exon 3 of the CTNNB1 gene (beta-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol 31: 248-255. doi: 10.1097/DAD.0b013e318198922a
|
| [207] |
Lu LC, Shao YY, Lee YH, et al. (2014) beta-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Oncology 87: 159-166. doi: 10.1159/000362821
|
| [208] | Hongyo T, Hoshida Y, Nakatsuka S, et al. (2005) p53, K-ras, c-kit and beta-catenin gene mutations in sinonasal NK/T-cell lymphoma in Korea and Japan. Oncol Rep 13: 265-271. |
| [209] | Kurniawan AN, Hongyo T, Hardjolukito ES, et al. (2006) Gene mutation analysis of sinonasal lymphomas in Indonesia. Oncol Rep 15: 1257-1263. |
| [210] | Garcia-Rostan G, Tallini G, Herrero A, et al. (1999) Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res 59: 1811-1815. |
| [211] | Sakamoto A, Oda Y, Adachi T, et al. (2002) Beta-catenin accumulation and gene mutation in exon 3 in dedifferentiated liposarcoma and malignant fibrous histiocytoma. Arch Pathol Lab Med 126: 1071-1078. |
| [212] |
Miyake T, Tanaka Y, Kato K, et al. (2006) Gene mutation analysis and immunohistochemical study of beta-catenin in odontogenic tumors. Pathol Int 56: 732-737. doi: 10.1111/j.1440-1827.2006.02039.x
|
| [213] |
Dutt A, Salvesen HB, Chen TH, et al. (2008) Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A 105: 8713-8717. doi: 10.1073/pnas.0803379105
|
| [214] |
Aitken SJ, Presneau N, Kalimuthu S, et al. (2015) Next-generation sequencing is highly sensitive for the detection of beta-catenin mutations in desmoid-type fibromatoses. Virchows Arch 467: 203-210. doi: 10.1007/s00428-015-1765-0
|
| [215] |
Matsumoto T, Yamazaki M, Takahashi H, et al. (2015) Distinct beta-catenin and PIK3CA mutation profiles in endometriosis-associated ovarian endometrioid and clear cell carcinomas. Am J Clin Pathol 144: 452-463. doi: 10.1309/AJCPZ5T2POOFMQVN
|
| [216] |
Shibata T, Kokubu A, Miyamoto M, et al. (2011) Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol 178: 1395-1402. doi: 10.1016/j.ajpath.2010.12.011
|
| [217] |
Hara K, Saito T, Hayashi T, et al. (2015) A mutation spectrum that includes GNAS, KRAS and TP53 may be shared by mucinous neoplasms of the appendix. Pathol Res Pract 211: 657-664. doi: 10.1016/j.prp.2015.06.004
|
| [218] |
Rogers HA, Miller S, Lowe J, et al. (2009) An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET). Br J Cancer 100: 1292-1302. doi: 10.1038/sj.bjc.6604979
|
| [219] |
Liu X, Qian Q, Xu P, et al. (2011) A novel conditionally replicating "armed" adenovirus selectively targeting gastrointestinal tumors with aberrant wnt signaling. Hum Gene Ther 22: 427-437. doi: 10.1089/hum.2010.128
|
| [220] |
Romero S, Szafranska J, Cabrera E, et al. (2012) Role of tumor-associated macrophages and angiogenesis in desmoid-type fibromatosis. Virchows Arch 461: 117-122. doi: 10.1007/s00428-012-1265-4
|
| [221] | Maiti S, Alam R, Amos CI, et al. (2000) Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60: 6288-6292. |
| [222] |
Fukuzawa R, Heathcott RW, Sano M, et al. (2004) Myogenesis in Wilms' tumors is associated with mutations of the WT1 gene and activation of Bcl-2 and the Wnt signaling pathway. Pediatr Dev Pathol 7: 125-137. doi: 10.1007/s10024-003-3023-8
|
| [223] |
Scott RH, Murray A, Baskcomb L, et al. (2012) Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3: 327-335. doi: 10.18632/oncotarget.468
|
| [224] |
Shimizu Y, Ikeda S, Fujimori M, et al. (2002) Frequent alterations in the Wnt signaling pathway in colorectal cancer with microsatellite instability. Genes Chromosomes Cancer 33: 73-81. doi: 10.1002/gcc.1226
|
| [225] |
Cieply B, Zeng G, Proverbs-Singh T, et al. (2009) Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology 49: 821-831. doi: 10.1002/hep.22695
|
| [226] |
Kawahara A, Harada H, Abe H, et al. (2011) Nuclear beta-catenin expression in basal cell adenomas of salivary gland. J Oral Pathol Med 40: 460-466. doi: 10.1111/j.1600-0714.2011.01010.x
|
| [227] | Mirabelli-Primdahl L, Gryfe R, Kim H, et al. (1999) Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res 59: 3346-3351. |
| [228] |
Saegusa M, Okayasu I (2001) Frequent nuclear beta-catenin accumulation and associated mutations in endometrioid-type endometrial and ovarian carcinomas with squamous differentiation. J Pathol 194: 59-67. doi: 10.1002/path.856
|
| [229] |
Schulze K, Imbeaud S, Letouze E, et al. (2015) Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47: 505-511. doi: 10.1038/ng.3252
|
| [230] |
Shinohara A, Yokoyama Y, Wan X, et al. (2001) Cytoplasmic/nuclear expression without mutation of exon 3 of the beta-catenin gene is frequent in the development of the neoplasm of the uterine cervix. Gynecol Oncol 82: 450-455. doi: 10.1006/gyno.2001.6298
|
| [231] |
Hodis E, Watson IR, Kryukov GV, et al. (2012) A landscape of driver mutations in melanoma. Cell 150: 251-263. doi: 10.1016/j.cell.2012.06.024
|
| [232] |
Groen RW, Oud ME, Schilder-Tol EJ, et al. (2008) Illegitimate WNT pathway activation by beta-catenin mutation or autocrine stimulation in T-cell malignancies. Cancer Res 68: 6969-6977. doi: 10.1158/0008-5472.CAN-08-1322
|
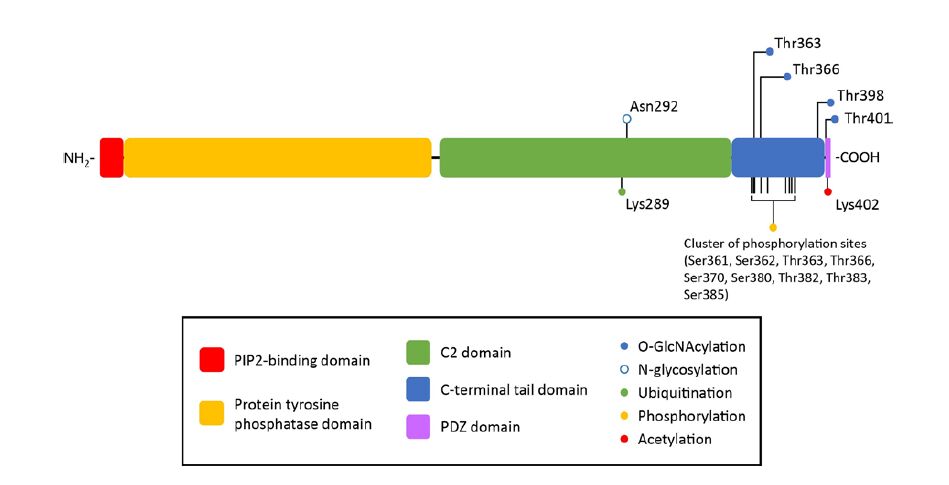
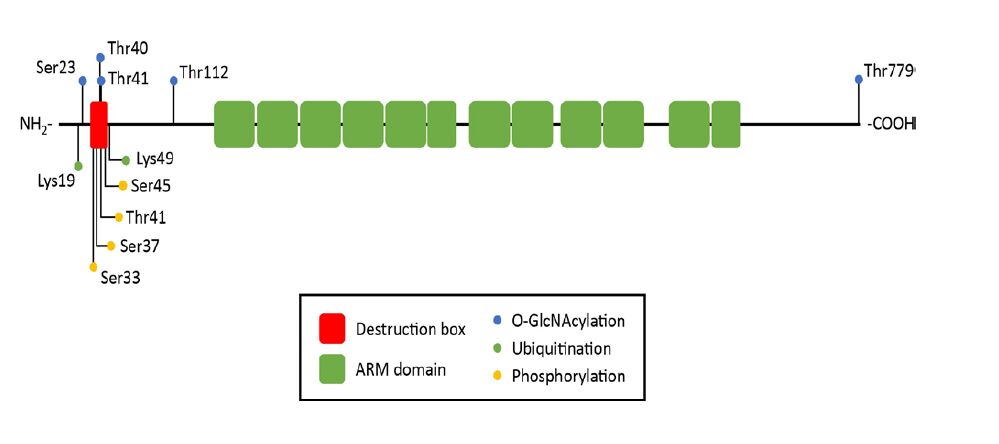
Figures(2) / Tables(3)

Carman K.M. Ip, Jun Yin, Patrick K.S. Ng, Shiaw-Yih Lin, Gordon B. Mills. Genomic-Glycosylation Aberrations in Tumor Initiation, Progression and Management[J]. AIMS Medical Science, 2016, 3(4): 386-416. doi: 10.3934/medsci.2016.4.386







 DownLoad:
DownLoad: