Citation: Angel Rivera-Calzada, Andrés López-Perrote, Roberto Melero, Jasminka Boskovic, Hugo Muñoz-Hernández, Fabrizio Martino, Oscar Llorca. Structure and Assembly of the PI3K-like Protein Kinases (PIKKs) Revealed by Electron Microscopy[J]. AIMS Biophysics, 2015, 2(2): 36-57. doi: 10.3934/biophy.2015.2.36

| [1] | Baretic D, Williams RL (2014) PIKKs - the solenoid nest where partners and kinases meet. Curr Opin Struct Biol 29C: 134-142. |
| [2] |
Lovejoy CA, Cortez D (2009) Common mechanisms of PIKK regulation. DNA Repair (Amst) 8: 1004-1008. doi: 10.1016/j.dnarep.2009.04.006

|
| [3] |
Lempiainen H, Halazonetis TD (2009) Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J 28: 3067-3073. doi: 10.1038/emboj.2009.281
|
| [4] |
van der Burg M, van Dongen JJ, van Gent DC (2009) DNA-PKcs deficiency in human: long predicted, finally found. Curr Opin Allergy Clin Immunol 9: 503-509. doi: 10.1097/ACI.0b013e3283327e41
|
| [5] |
Savitsky K, Bar-Shira A, Gilad S, et al. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749-1753. doi: 10.1126/science.7792600
|
| [6] | Weber AM, Ryan AJ (2014) ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. |
| [7] |
Roberts TL, Ho U, Luff J, et al. (2013) Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc Natl Acad Sci U S A 110: E285-294. doi: 10.1073/pnas.1215696110
|
| [8] |
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21-35. doi: 10.1038/nrm3025
|
| [9] |
Kong X, Shen Y, Jiang N, et al. (2011) Emerging roles of DNA-PK besides DNA repair. Cell Signal 23: 1273-1280. doi: 10.1016/j.cellsig.2011.04.005
|
| [10] | Kruger A, Ralser M (2011) ATM is a redox sensor linking genome stability and carbon metabolism. Sci Signal 4: pe17. |
| [11] |
Oliveira V, Romanow WJ, Geisen C, et al. (2008) A protective role for the human SMG-1 kinase against tumor necrosis factor-alpha-induced apoptosis. J Biol Chem 283: 13174-13184. doi: 10.1074/jbc.M708008200
|
| [12] |
Bosotti R, Isacchi A, Sonnhammer EL (2000) FAT: a novel domain in PIK-related kinases. Trends Biochem Sci 25: 225-227. doi: 10.1016/S0968-0004(00)01563-2
|
| [13] |
Keith CT, Schreiber SL (1995) PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 270: 50-51. doi: 10.1126/science.270.5233.50
|
| [14] |
Brewerton SC, Dore AS, Drake AC, et al. (2004) Structural analysis of DNA-PKcs: modelling of the repeat units and insights into the detailed molecular architecture. J Struct Biol 145: 295-306. doi: 10.1016/j.jsb.2003.11.024
|
| [15] |
Perry J, Kleckner N (2003) The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 112: 151-155. doi: 10.1016/S0092-8674(03)00033-3
|
| [16] |
Sommer LA, Schaad M, Dames SA (2013) NMR- and circular dichroism-monitored lipid binding studies suggest a general role for the FATC domain as membrane anchor of phosphatidylinositol 3-kinase-related kinases (PIKK). J Biol Chem 288: 20046-20063. doi: 10.1074/jbc.M113.467233
|
| [17] |
Lucero H, Gae D, Taccioli GE (2003) Novel localization of the DNA-PK complex in lipid rafts: a putative role in the signal transduction pathway of the ionizing radiation response. J Biol Chem 278: 22136-22143. doi: 10.1074/jbc.M301579200
|
| [18] |
Dames SA (2010) Structural basis for the association of the redox-sensitive target of rapamycin FATC domain with membrane-mimetic micelles. J Biol Chem 285: 7766-7775. doi: 10.1074/jbc.M109.058404
|
| [19] |
Sancak Y, Bar-Peled L, Zoncu R, et al. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290-303. doi: 10.1016/j.cell.2010.02.024
|
| [20] |
Morita T, Yamashita A, Kashima I, et al. (2007) Distant N- and C-terminal domains are required for intrinsic kinase activity of SMG-1, a critical component of nonsense-mediated mRNA decay. J Biol Chem 282: 7799-7808. doi: 10.1074/jbc.M610159200
|
| [21] |
Yang H, Rudge DG, Koos JD, et al. (2013) mTOR kinase structure, mechanism and regulation. Nature 497: 217-223. doi: 10.1038/nature12122
|
| [22] | Sirbu BM, Cortez D (2013) DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5: a012724. |
| [23] |
Ochi T, Wu Q, Blundell TL (2014) The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair (Amst) 17: 98-109. doi: 10.1016/j.dnarep.2014.02.010
|
| [24] |
Spagnolo L, Rivera-Calzada A, Pearl LH, et al. (2006) Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol Cell 22: 511-519. doi: 10.1016/j.molcel.2006.04.013
|
| [25] |
Hammel M, Yu Y, Mahaney BL, et al. (2010) Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem 285: 1414-1423. doi: 10.1074/jbc.M109.065615
|
| [26] |
Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308: 551-554. doi: 10.1126/science.1108297
|
| [27] |
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: 1542-1548. doi: 10.1126/science.1083430
|
| [28] |
Heitman J, Movva NR, Hall MN (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253: 905-909. doi: 10.1126/science.1715094
|
| [29] |
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149: 274-293. doi: 10.1016/j.cell.2012.03.017
|
| [30] |
Shimobayashi M, Hall MN (2014) Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 15: 155-162. doi: 10.1038/nrm3757
|
| [31] |
Yamashita A (2013) Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells 18: 161-175. doi: 10.1111/gtc.12033
|
| [32] |
Yamashita A, Izumi N, Kashima I, et al. (2009) SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev 23: 1091-1105. doi: 10.1101/gad.1767209
|
| [33] |
Ivanov PV, Gehring NH, Kunz JB, et al. (2008) Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. Embo J 27: 736-747. doi: 10.1038/emboj.2008.17
|
| [34] |
Kashima I, Yamashita A, Izumi N, et al. (2006) Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20: 355-367. doi: 10.1101/gad.1389006
|
| [35] |
Murr R, Vaissiere T, Sawan C, et al. (2007) Orchestration of chromatin-based processes: mind the TRRAP. Oncogene 26: 5358-5372. doi: 10.1038/sj.onc.1210605
|
| [36] |
McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556-562. doi: 10.1128/MCB.20.2.556-562.2000
|
| [37] |
Sibanda BL, Chirgadze DY, Blundell TL (2010) Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463: 118-121. doi: 10.1038/nature08648
|
| [38] |
Stark H (2010) GraFix: stabilization of fragile macromolecular complexes for single particle cryo-EM. Methods Enzymol 481: 109-126. doi: 10.1016/S0076-6879(10)81005-5
|
| [39] |
Boskovic J, Rivera-Calzada A, Maman JD, et al. (2003) Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J 22: 5875-5882. doi: 10.1093/emboj/cdg555
|
| [40] |
Melero R, Uchiyama A, Castano R, et al. (2014) Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure 22: 1105-1119. doi: 10.1016/j.str.2014.05.015
|
| [41] |
Dames SA, Mulet JM, Rathgeb-Szabo K, et al. (2005) The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J Biol Chem 280: 20558-20564. doi: 10.1074/jbc.M501116200
|
| [42] | Leone M, Crowell KJ, Chen J, et al. (2006) The FRB domain of mTOR: NMR solution structure and inhibitor design. Biochemistry 45: 10294-10302. |
| [43] |
Yip CK, Murata K, Walz T, et al. (2010) Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell 38: 768-774. doi: 10.1016/j.molcel.2010.05.017
|
| [44] |
Williams DR, Lee KJ, Shi J, et al. (2008) Cryo-EM structure of the DNA-dependent protein kinase catalytic subunit at subnanometer resolution reveals alpha helices and insight into DNA binding. Structure 16: 468-477. doi: 10.1016/j.str.2007.12.014
|
| [45] |
Rivera-Calzada A, Maman JD, Spagnolo L, et al. (2005) Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure 13: 243-255. doi: 10.1016/j.str.2004.12.006
|
| [46] | Kuhlbrandt W (2014) Cryo-EM enters a new era. Elife 3: e03678. |
| [47] |
Llorca O, Rivera-Calzada A, Grantham J, et al. (2003) Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene 22: 3867-3874. doi: 10.1038/sj.onc.1206649
|
| [48] |
Arias-Palomo E, Yamashita A, Fernandez IS, et al. (2011) The nonsense-mediated mRNA decay SMG-1 kinase is regulated by large-scale conformational changes controlled by SMG-8. Genes Dev 25: 153-164. doi: 10.1101/gad.606911
|
| [49] |
Adami A, Garcia-Alvarez B, Arias-Palomo E, et al. (2007) Structure of TOR and its complex with KOG1. Mol Cell 27: 509-516. doi: 10.1016/j.molcel.2007.05.040
|
| [50] |
Chiu CY, Cary RB, Chen DJ, et al. (1998) Cryo-EM imaging of the catalytic subunit of the DNA-dependent protein kinase. J Mol Biol 284: 1075-1081. doi: 10.1006/jmbi.1998.2212
|
| [51] |
Leuther KK, Hammarsten O, Kornberg RD, et al. (1999) Structure of DNA-dependent protein kinase: implications for its regulation by DNA. EMBO J 18: 1114-1123. doi: 10.1093/emboj/18.5.1114
|
| [52] |
Grinthal A, Adamovic I, Weiner B, et al. (2010) PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc Natl Acad Sci USA 107: 2467-2472. doi: 10.1073/pnas.0914073107
|
| [53] |
Forwood JK, Lange A, Zachariae U, et al. (2010) Quantitative structural analysis of importin-beta flexibility: paradigm for solenoid protein structures. Structure 18: 1171-1183. doi: 10.1016/j.str.2010.06.015
|
| [54] |
Knutson BA (2010) Insights into the domain and repeat architecture of target of rapamycin. J Struct Biol 170: 354-363. doi: 10.1016/j.jsb.2010.01.002
|
| [55] |
Spagnolo L, Barbeau J, Curtin NJ, et al. (2012) Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res 40: 4168-4177. doi: 10.1093/nar/gkr1231
|
| [56] |
Morris EP, Rivera-Calzada A, da Fonseca PC, et al. (2011) Evidence for a remodelling of DNA-PK upon autophosphorylation from electron microscopy studies. Nucleic Acids Res 39: 5757-5767. doi: 10.1093/nar/gkr146
|
| [57] |
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499-506. doi: 10.1038/nature01368
|
| [58] | Perry JJ, Tainer JA (2011) All stressed out without ATM kinase. Sci Signal 4: pe18. |
| [59] |
Guo Z, Kozlov S, Lavin MF, et al. (2010) ATM activation by oxidative stress. Science 330: 517-521. doi: 10.1126/science.1192912
|
| [60] |
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197-210. doi: 10.1038/nrm3546
|
| [61] |
Dynan WS, Yoo S (1998) Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res 26: 1551-1559. doi: 10.1093/nar/26.7.1551
|
| [62] |
Neal JA, Sugiman-Marangos S, VanderVere-Carozza P, et al. (2014) Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol Cell Biol 34: 2162-2175. doi: 10.1128/MCB.01554-13
|
| [63] |
Meek K, Douglas P, Cui X, et al. (2007) trans Autophosphorylation at DNA-dependent protein kinase's two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol 27: 3881-3890. doi: 10.1128/MCB.02366-06
|
| [64] |
Dobbs TA, Tainer JA, Lees-Miller SP (2010) A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst) 9: 1307-1314. doi: 10.1016/j.dnarep.2010.09.019
|
| [65] |
Villarreal SA, Stewart PL (2014) CryoEM and image sorting for flexible protein/DNA complexes. J Struct Biol 187: 76-83. doi: 10.1016/j.jsb.2013.12.002
|
| [66] | Wu PY, Ruhlmann C, Winston F, et al. (2004) Molecular architecture of the S. cerevisiae SAGA complex. Mol Cell 15: 199-208. |
| [67] |
Chittuluru JR, Chaban Y, Monnet-Saksouk J, et al. (2011) Structure and nucleosome interaction of the yeast NuA4 and Piccolo-NuA4 histone acetyltransferase complexes. Nat Struct Mol Biol 18: 1196-1203. doi: 10.1038/nsmb.2128
|
| [68] |
Kozlov SV, Graham ME, Jakob B, et al. (2011) Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem 286: 9107-9119. doi: 10.1074/jbc.M110.204065
|
| [69] |
Zhao R, Davey M, Hsu YC, et al. (2005) Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 120: 715-727. doi: 10.1016/j.cell.2004.12.024
|
| [70] |
Boulon S, Bertrand E, Pradet-Balade B (2012) HSP90 and the R2TP co-chaperone complex: building multi-protein machineries essential for cell growth and gene expression. RNA Biol 9: 148-154. doi: 10.4161/rna.18494
|
| [71] |
Hurov KE, Cotta-Ramusino C, Elledge SJ (2010) A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 24: 1939-1950. doi: 10.1101/gad.1934210
|
| [72] |
Takai H, Wang RC, Takai KK, et al. (2007) Tel2 regulates the stability of PI3K-related protein kinases. Cell 131: 1248-1259. doi: 10.1016/j.cell.2007.10.052
|
| [73] |
Horejsi Z, Takai H, Adelman CA, et al. (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell 39: 839-850. doi: 10.1016/j.molcel.2010.08.037
|
| [74] | Izumi N, Yamashita A, Iwamatsu A, et al. (2010) AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci Signal 3: ra27. |
| [75] |
Pal M, Morgan M, Phelps SE, et al. (2014) Structural basis for phosphorylation-dependent recruitment of Tel2 to Hsp90 by Pih1. Structure 22: 805-818. doi: 10.1016/j.str.2014.04.001
|
| [76] |
Torreira E, Jha S, Lopez-Blanco JR, et al. (2008) Architecture of the pontin/reptin complex, essential in the assembly of several macromolecular complexes. Structure 16: 1511-1520. doi: 10.1016/j.str.2008.08.009
|
| [77] |
Lopez-Perrote A, Munoz-Hernandez H, Gil D, et al. (2012) Conformational transitions regulate the exposure of a DNA-binding domain in the RuvBL1-RuvBL2 complex. Nucleic Acids Res 40: 11086-11099. doi: 10.1093/nar/gks871
|
| [78] |
Matias PM, Gorynia S, Donner P, et al. (2006) Crystal structure of the human AAA+ protein RuvBL1. J Biol Chem 281: 38918-38929. doi: 10.1074/jbc.M605625200
|
| [79] |
Gorynia S, Bandeiras TM, Pinho FG, et al. (2011) Structural and functional insights into a dodecameric molecular machine - the RuvBL1/RuvBL2 complex. J Struct Biol 176: 279-291. doi: 10.1016/j.jsb.2011.09.001
|
| [80] |
Huen J, Kakihara Y, Ugwu F, et al. (2010) Rvb1-Rvb2: essential ATP-dependent helicases for critical complexes. Biochem Cell Biol 88: 29-40. doi: 10.1139/O09-122
|
| [81] |
Tosi A, Haas C, Herzog F, et al. (2013) Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell 154: 1207-1219. doi: 10.1016/j.cell.2013.08.016
|
| [82] |
Nguyen VQ, Ranjan A, Stengel F, et al. (2013) Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. Cell 154: 1220-1231. doi: 10.1016/j.cell.2013.08.018
|
| [83] |
Jha S, Dutta A (2009) RVB1/RVB2: running rings around molecular biology. Mol Cell 34: 521-533. doi: 10.1016/j.molcel.2009.05.016
|
| [84] |
Melero R, Buchwald G, Castano R, et al. (2012) The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3' end. Nat Struct Mol Biol 19: 498-505, S491-492. doi: 10.1038/nsmb.2287
|
| [85] |
Chakrabarti S, Jayachandran U, Bonneau F, et al. (2011) Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41: 693-703. doi: 10.1016/j.molcel.2011.02.010
|
| [86] |
Ali MM, Roe SM, Vaughan CK, et al. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440: 1013-1017. doi: 10.1038/nature04716
|
| [87] |
Takai H, Xie Y, de Lange T, et al. (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24: 2019-2030. doi: 10.1101/gad.1956410
|
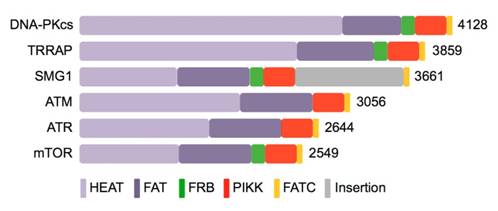
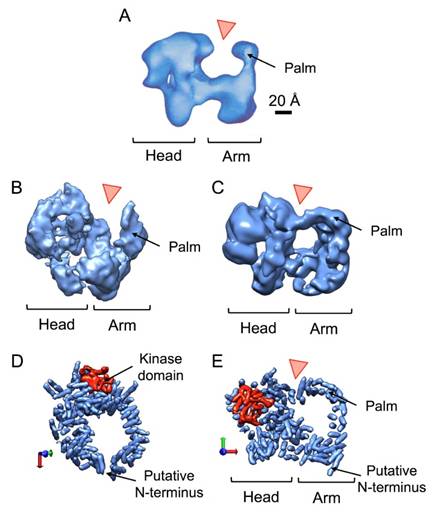
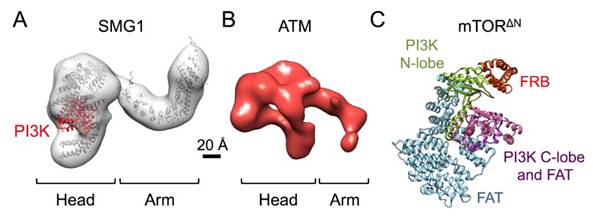
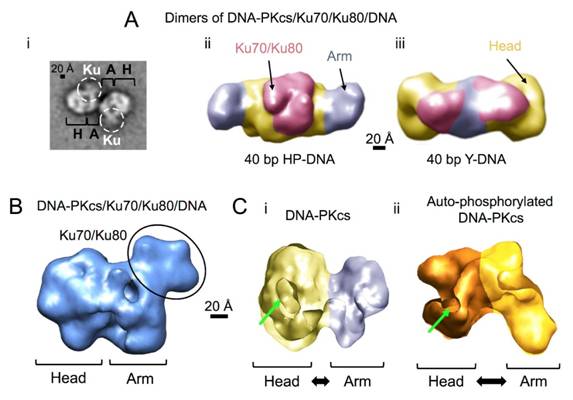
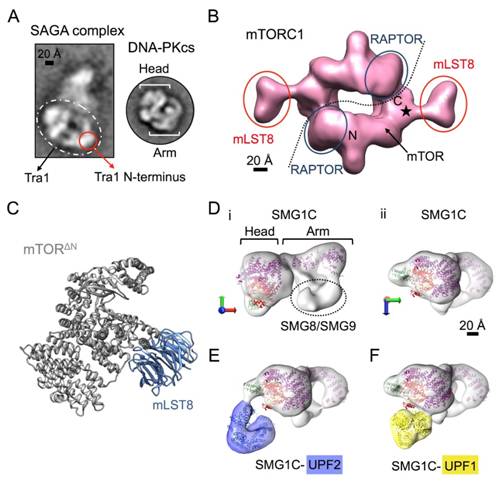
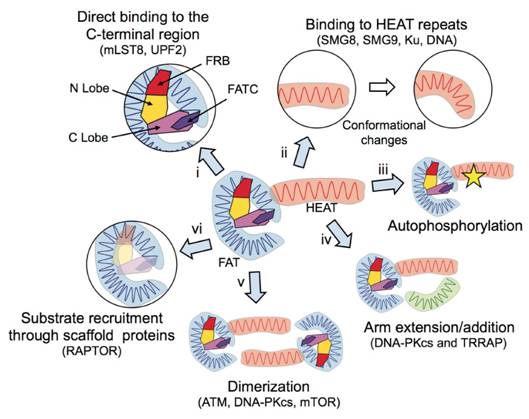
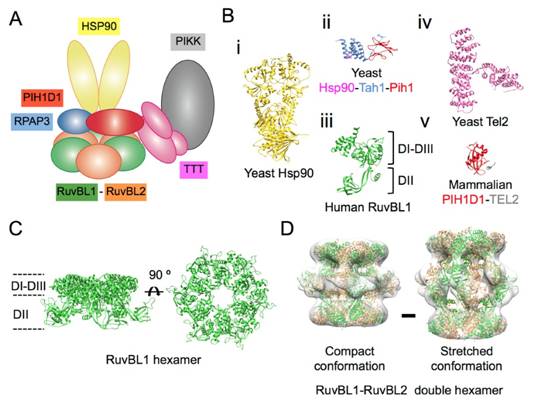
Figures(7)

Angel Rivera-Calzada, Andrés López-Perrote, Roberto Melero, Jasminka Boskovic, Hugo Muñoz-Hernández, Fabrizio Martino, Oscar Llorca. Structure and Assembly of the PI3K-like Protein Kinases (PIKKs) Revealed by Electron Microscopy[J]. AIMS Biophysics, 2015, 2(2): 36-57. doi: 10.3934/biophy.2015.2.36







 DownLoad:
DownLoad: