Citation: J Bernard Heymann. Validation of 3D EM Reconstructions: The Phantom in the Noise[J]. AIMS Biophysics, 2015, 2(1): 21-35. doi: 10.3934/biophy.2015.1.21

| [1] | Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343: 1443–1444. |
| [2] |
Henderson R, Sali A, Baker ML, et al. (2012) Outcome of the first electron microscopy validation task force meeting. Structure 20: 205–214. doi: 10.1016/j.str.2011.12.014

|
| [3] | Harauz G, van Heel M (1986) Exact filters for general geometry three dimensional reconstruction. Optik 73: 146–156. |
| [4] |
van Heel M (2013) Finding trimeric HIV-1 envelope glycoproteins in random noise. Proc Natl Acad Sci U S A 110: E4175–4177. doi: 10.1073/pnas.1314353110
|
| [5] |
Brunger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355: 472–475. doi: 10.1038/355472a0
|
| [6] |
Baker TS, Cheng RH (1996) A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. Journal of Structural Biology 116:120–130. doi: 10.1006/jsbi.1996.0020
|
| [7] |
Scheres SH, Chen S (2012) Prevention of overfitting in cryo-EM structure determination. Nature methods 9: 853–854. doi: 10.1038/nmeth.2115
|
| [8] |
Henderson R, Chen S, Chen JZ, et al. (2011) Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol 413: 1028–1046. doi: 10.1016/j.jmb.2011.09.008
|
| [9] |
Heymann JB, Belnap DM (2007) Bsoft: Image processing and molecular modeling for electron microscopy. J Struct Biol 157: 3–18. doi: 10.1016/j.jsb.2006.06.006
|
| [10] |
Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333: 721–745. doi: 10.1016/j.jmb.2003.07.013
|
| [11] |
Shaikh TR, Hegerl R, Frank J (2003) An approach to examining model dependence in EM reconstructions using cross-validation. J Struct Biol 142: 301–310. doi: 10.1016/S1047-8477(03)00029-7
|
| [12] |
Falkner B, Schroder GF (2013) Cross-validation in cryo-EM-based structural modeling. Proc Natl Acad Sci U S A 110: 8930–8935. doi: 10.1073/pnas.1119041110
|
| [13] |
Chen S, McMullan G, Faruqi AR, et al. (2013) High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135: 24–35. doi: 10.1016/j.ultramic.2013.06.004
|
| [14] |
DiMaio F, Zhang J, Chiu W, et al. (2013) Cryo-EM model validation using independent map reconstructions. Protein Sci 22: 865–868. doi: 10.1002/pro.2267
|
| [15] |
Belnap DM, Olson NH, Baker TS (1997) A method for establishing the handedness of biological macromolecules. J Struct Biol 120: 44–51. doi: 10.1006/jsbi.1997.3896
|
| [16] |
Henderson R (2013) Avoiding the pitfalls of single particle cryo-electron microscopy: Einstein from noise. Proc Natl Acad Sci U S A 110: 18037–18041. doi: 10.1073/pnas.1314449110
|
| [17] |
Mao Y, Castillo-Menendez LR, Sodroski JG (2013) Reply to Subramaniam, van Heel, and Henderson: Validity of the cryo-electron microscopy structures of the HIV-1 envelope glycoprotein complex. Proc Natl Acad Sci U S A 110: E4178–4182. doi: 10.1073/pnas.1316666110
|
| [18] |
Mao Y, Wang L, Gu C, et al. (2013) Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc Natl Acad Sci U S A 110: 12438–12443. doi: 10.1073/pnas.1307382110
|
| [19] | Subramaniam S (2013) Structure of trimeric HIV-1 envelope glycoproteins. Proc Natl Acad Sci110: E4172–E4174. |
| [20] |
Glaeser RM (2013) Replication and validation of cryo-EM structures. J Struct Biol 184:379–380. doi: 10.1016/j.jsb.2013.09.007
|
| [21] | Patwardhan A, Carazo JM, Carragher B, et al. (2012) Data management challenges in three-dimensional EM. Nature structural & molecular biology 19: 1203–1207. |
| [22] |
Larson SB, Day JS, Nguyen C, et al. (2009) High-resolution structure of proteinase K cocrystallized with digalacturonic acid. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:192–198. doi: 10.1107/S1744309109002218
|
| [23] |
Zhang X, Meining W, Cushman M, et al. (2003) A structure-based model of the reaction catalyzed by lumazine synthase from Aquifex aeolicus. J Mol Biol 328: 167–182. doi: 10.1016/S0022-2836(03)00186-4
|
| [24] |
Gatsogiannis C, Lang AE, Meusch D, et al. (2013) A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature 495: 520–523. doi: 10.1038/nature11987
|
| [25] |
Liu X, Zhang Q, Murata K, et al. (2010) Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat Struct Mol Biol 17: 830–836. doi: 10.1038/nsmb.1823
|
| [26] |
Jensen GJ (2001) Alignment error envelopes for single particle analysis. J Struct Biol 133:143–155. doi: 10.1006/jsbi.2001.4334
|
| [27] | Baldwin PR, Penczek PA (2005) Estimating alignment errors in sets of 2-D images. J Struct Biol150: 211–225. |
| [28] |
Joyeux L, Penczek PA (2002) Efficiency of 2D alignment methods. Ultramicroscopy 92: 33–46. doi: 10.1016/S0304-3991(01)00154-1
|
| [29] |
Gatsogiannis C, Markl J (2009) Keyhole limpet hemocyanin: 9-A CryoEM structure and molecular model of the KLH1 didecamer reveal the interfaces and intricate topology of the 160 functional units. J Mol Biol 385: 963–983. doi: 10.1016/j.jmb.2008.10.080
|
| [30] |
Van Heel M (1982) Detection of objects in quantum-noise-limited images. Ultramicroscopy 7: 331–341. doi: 10.1016/0304-3991(82)90258-3
|
| [31] |
Unser M, Sorzano CO, Thevenaz P, et al. (2005) Spectral signal-to-noise ratio and resolution assessment of 3D reconstructions. J Struct Biol 149: 243–255. doi: 10.1016/j.jsb.2004.10.011
|
| [32] |
Liao HY, Frank J (2010) Definition and estimation of resolution in single-particle reconstructions. Structure 18: 768–775. doi: 10.1016/j.str.2010.05.008
|
| [33] |
Baxter WT, Grassucci RA, Gao H, et al. (2009) Determination of signal-to-noise ratios and spectral SNRs in cryo-EM low-dose imaging of molecules. J Struct Biol 166: 126–132. doi: 10.1016/j.jsb.2009.02.012
|
| [34] | Borgnia MJ, Shi D, Zhang P, et al. (2004) Visualization of alpha-helical features in a density map constructed using 9 molecular images of the 1.8 MDa icosahedral core of pyruvate dehydrogenase. J Struct Biol 147: 136–145. |
| [35] |
Liu X, Jiang W, Jakana J, et al. (2007) Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. J Struct Biol 160: 11–27. doi: 10.1016/j.jsb.2007.06.009
|
| [36] | LeBarron J, Grassucci RA, Shaikh TR, et al. (2008) Exploration of parameters in cryo-EM leading to an improved density map of the E. coli ribosome. J Struct Biol 164: 24–32. |
| [37] |
Stagg SM, Lander GC, Quispe J, et al. (2008) A test-bed for optimizing high-resolution single particle reconstructions. J Struct Biol 163: 29–39. doi: 10.1016/j.jsb.2008.04.005
|
| [38] |
Stagg SM, Noble AJ, Spilman M, et al. (2014) ResLog plots as an empirical metric of the quality of cryo-EM reconstructions. J Struct Biol 185: 418–426. doi: 10.1016/j.jsb.2013.12.010
|
| [39] |
Cardone G, Yan X, Sinkovits RS, et al. (2013) Three-dimensional reconstruction of icosahedral particles from single micrographs in real time at the microscope. J Struct Biol 183: 329–341. doi: 10.1016/j.jsb.2013.07.007
|
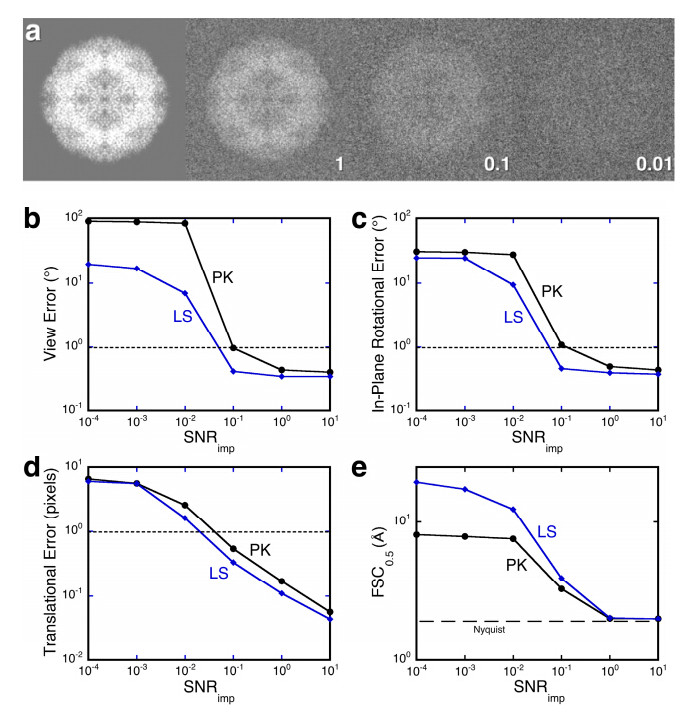
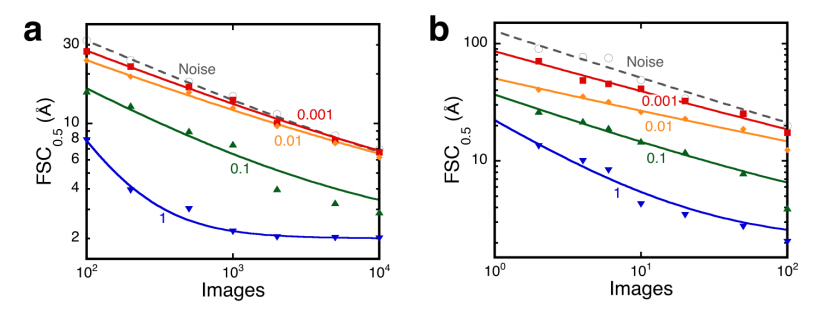
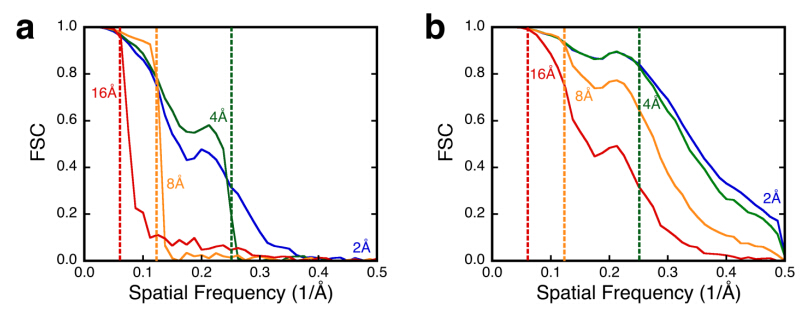
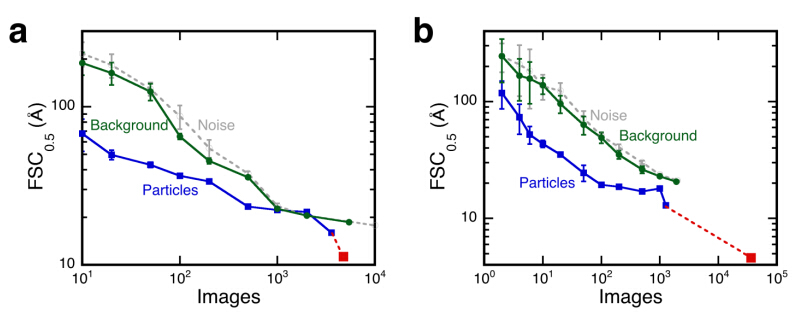
Figures(5)

J Bernard Heymann. Validation of 3D EM Reconstructions: The Phantom in the Noise[J]. AIMS Biophysics, 2015, 2(1): 21-35. doi: 10.3934/biophy.2015.1.21







 DownLoad:
DownLoad: