Citation: Warin Rangubpit, Pornthep Sompornpisut, R.B. Pandey. Thermal-induced unfolding-refolding of a nucleocapsid COVN protein[J]. AIMS Biophysics, 2021, 8(1): 103-110. doi: 10.3934/biophy.2021007

| [1] | Bzówka M, Mitusinska K, Raczynska A, et al. (2020) Molecular dynamics simulations indicate the COVID-19 mpro is not a viable target for small-molecule inhibitors design. bioRxiv doi.org/10.1101/2020.02.27.968008. |
| [2] |
J Alsaadi EA, Jones IM (2019) Membrane binding proteins of coronaviruses. Future Virol 14: 275-286. doi: 10.2217/fvl-2018-0144

|
| [3] | Wan Y, Shang J, Graham R, et al. (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94: e00127. |
| [4] |
Xu J, Zhao S, Teng T, et al. (2020) Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12: 244. doi: 10.3390/v12020244
|
| [5] |
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12: 254. doi: 10.3390/v12030254
|
| [6] | UniProtKB-P59595(NCAP_SARS) (2020) .Available from: https://www.uniprot.org/uniprot/P59595. |
| [7] |
Chang CK, Hsu YL, Chang YH, et al. (2009) Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J Virol 83: 2255-2264. doi: 10.1128/JVI.02001-08
|
| [8] |
Fang HJ, Chen YZ, Li MS, et al. (2009) Thermostability of the N-terminal RNA-binding domain of the SARS-CoV nucleocapsid protein: Experiments and numerical simulations. Biophys J 96: 1892-1901. doi: 10.1016/j.bpj.2008.10.045
|
| [9] |
Chang C, Chen CMM, Chiang M, et al. (2013) Transient oligomerization of the SARS-CoV N protein–implication for virus ribonucleoprotein packaging. PloS One 8: e65045. doi: 10.1371/journal.pone.0065045
|
| [10] |
Sorokina I, Mushegian A (2018) Modeling protein folding in vivo. Biol Direct 13: 13. doi: 10.1186/s13062-018-0217-6
|
| [11] |
Gershenson A, Gosavi S, Faccioli P, et al. (2020) Successes and challenges in simulating the folding of large proteins. J Biol Chem 295: 15-33. doi: 10.1074/jbc.REV119.006794
|
| [12] |
Miyazawa S, Jernigan RL (1985) Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 18: 534-552. doi: 10.1021/ma00145a039
|
| [13] |
Miyazawa S, Jernigan RL (1996) Residue–residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol 256: 623-644. doi: 10.1006/jmbi.1996.0114
|
| [14] |
Betancourt MR, Thirumalai D (1999) Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci 8: 361-369. doi: 10.1110/ps.8.2.361
|
| [15] |
Tanaka S, Scheraga HA (1976) Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules 9: 945-950. doi: 10.1021/ma60054a013
|
| [16] |
Godzik A (1996) Knowledge-based potentials for protein folding: what can we learn from known protein structures? Structure 4: 363-366. doi: 10.1016/S0969-2126(96)00041-X
|
| [17] |
Huang SY, Zou X (2011) Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins: Struct, Funct, Bioinf 79: 2648-2661. doi: 10.1002/prot.23086
|
| [18] |
Kitjaruwankul S, Khrutto C, Sompornpisut P, et al. (2016) Asymmetry in structural response of inner and outer transmembrane segments of CorA protein by a coarse-grain model. J Chem Phys 145: 135101. doi: 10.1063/1.4963807
|
| [19] |
Boonamnaj P, Paudel SS, Jetsadawisut W, et al. (2019) Thermal-response of a protein (hHv1) by a coarse-grained MC and all-atom MD computer simulations. Phys A 527: 121310. doi: 10.1016/j.physa.2019.121310
|
| [20] |
Fritsche M, Pandey RB, Farmer BL, et al. (2013) Variation in structure of a protein (H2AX) with knowledge-based interactions. PLoS One 8: e64507. doi: 10.1371/journal.pone.0064507
|
| [21] |
Pandey RB, Farmer BL, Gerstman BS (2015) Self-assembly dynamics for the transition of a globular aggregate to a fibril network of lysozyme proteins via a coarse-grained Monte Carlo simulation. AIP Adv 5: 092502. doi: 10.1063/1.4921074
|
| [22] |
Mirau P, Farmer BL, Pandey RB (2015) Structural variation of alpha-synuclein with temperature by a coarse-grained approach with knowledge-based interactions. AIP Adv 5: 092504. doi: 10.1063/1.4927544
|
| [23] |
Pokhrel R, Sompornpisut P, Chapagain P, et al. (2018) Domain rearrangement and denaturation in Ebola virus protein VP40. AIP Adv 8: 125129. doi: 10.1063/1.5063474
|
| [24] |
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16: 69. doi: 10.1186/s12985-019-1182-0
|
| [25] | Pandey RB Thermal denaturation of a protein (CoVE) by a coarse-grained Monte Carlo simulation (2020) .arXiv preprint arXiv:2009.00049, 2020. |
 biophy-08-01-007-s001.pdf biophy-08-01-007-s001.pdf |
 |
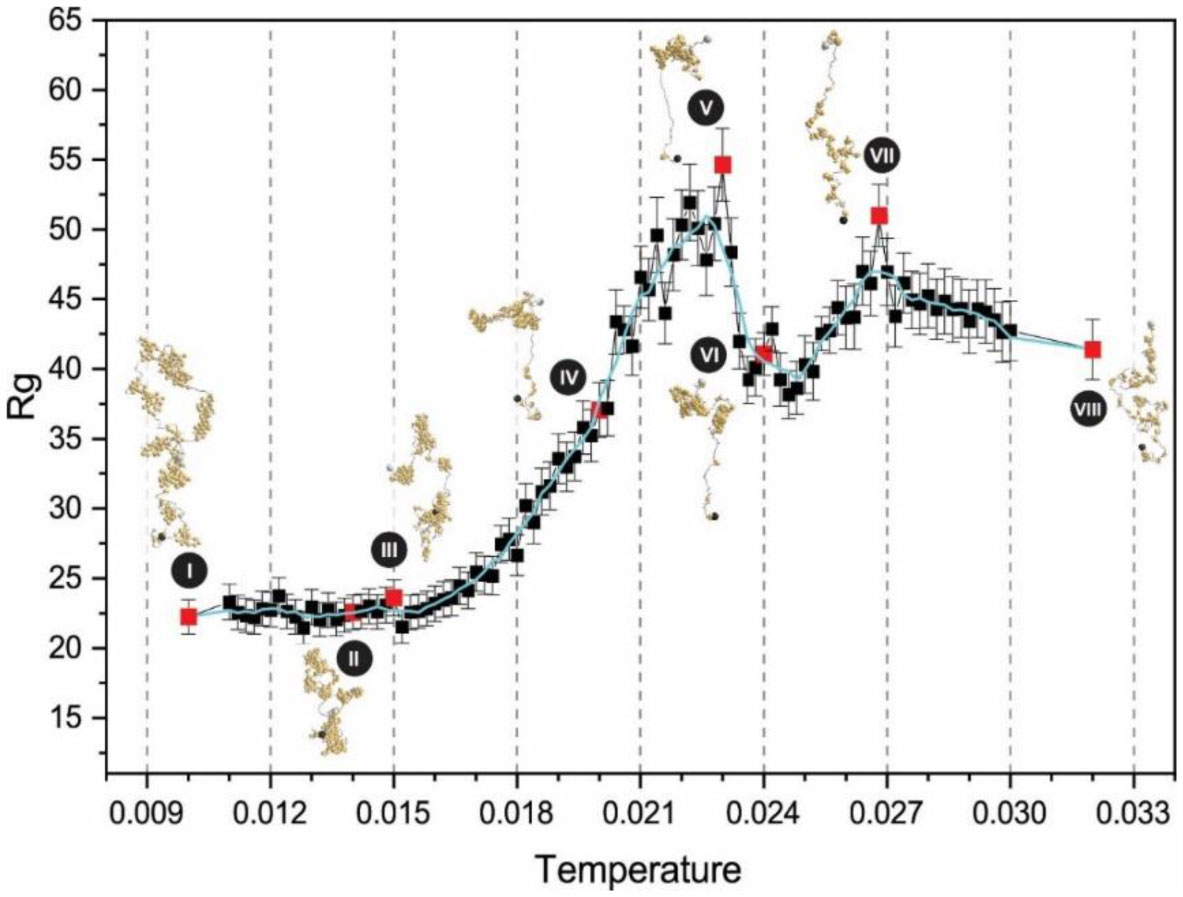
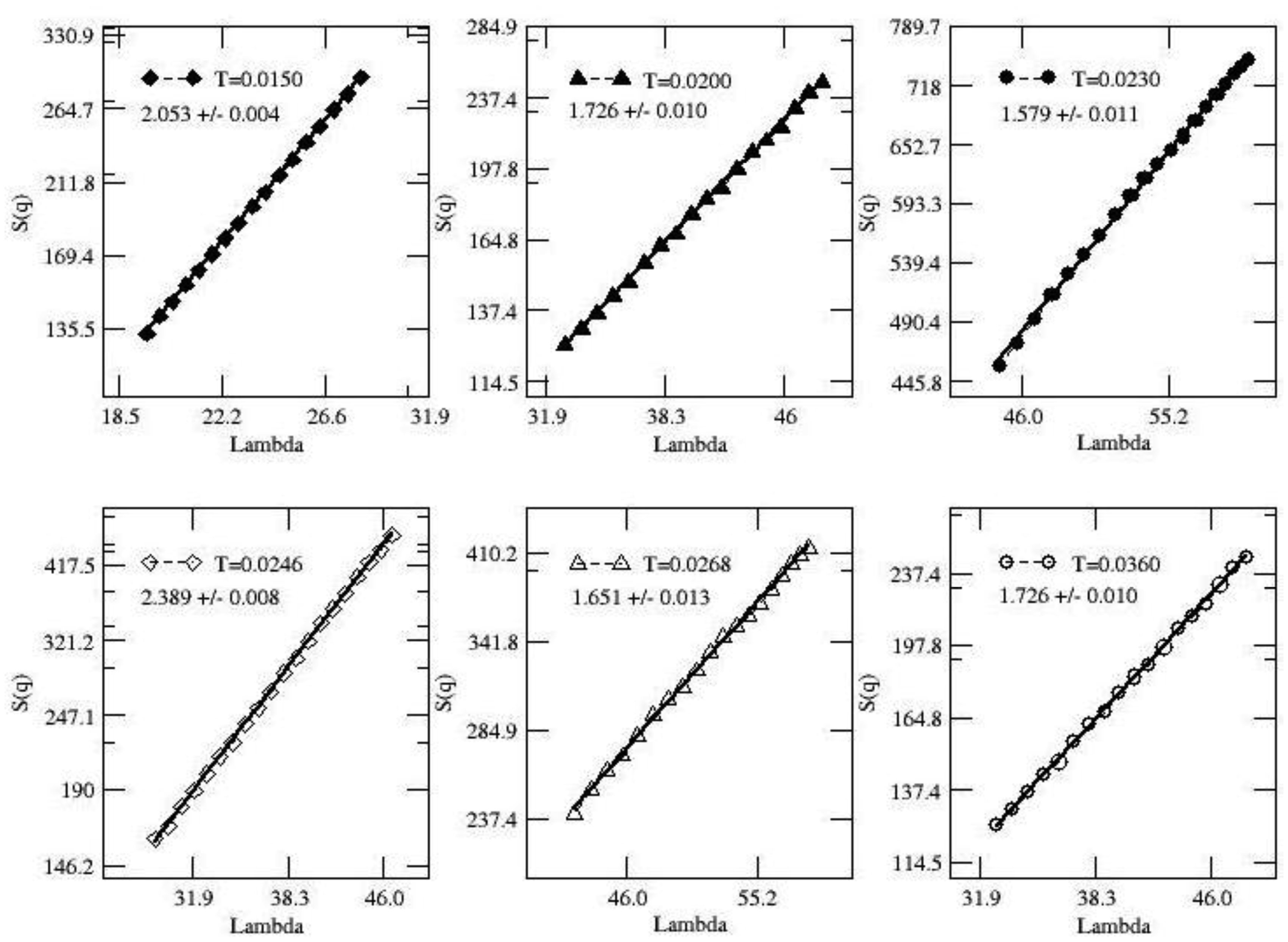
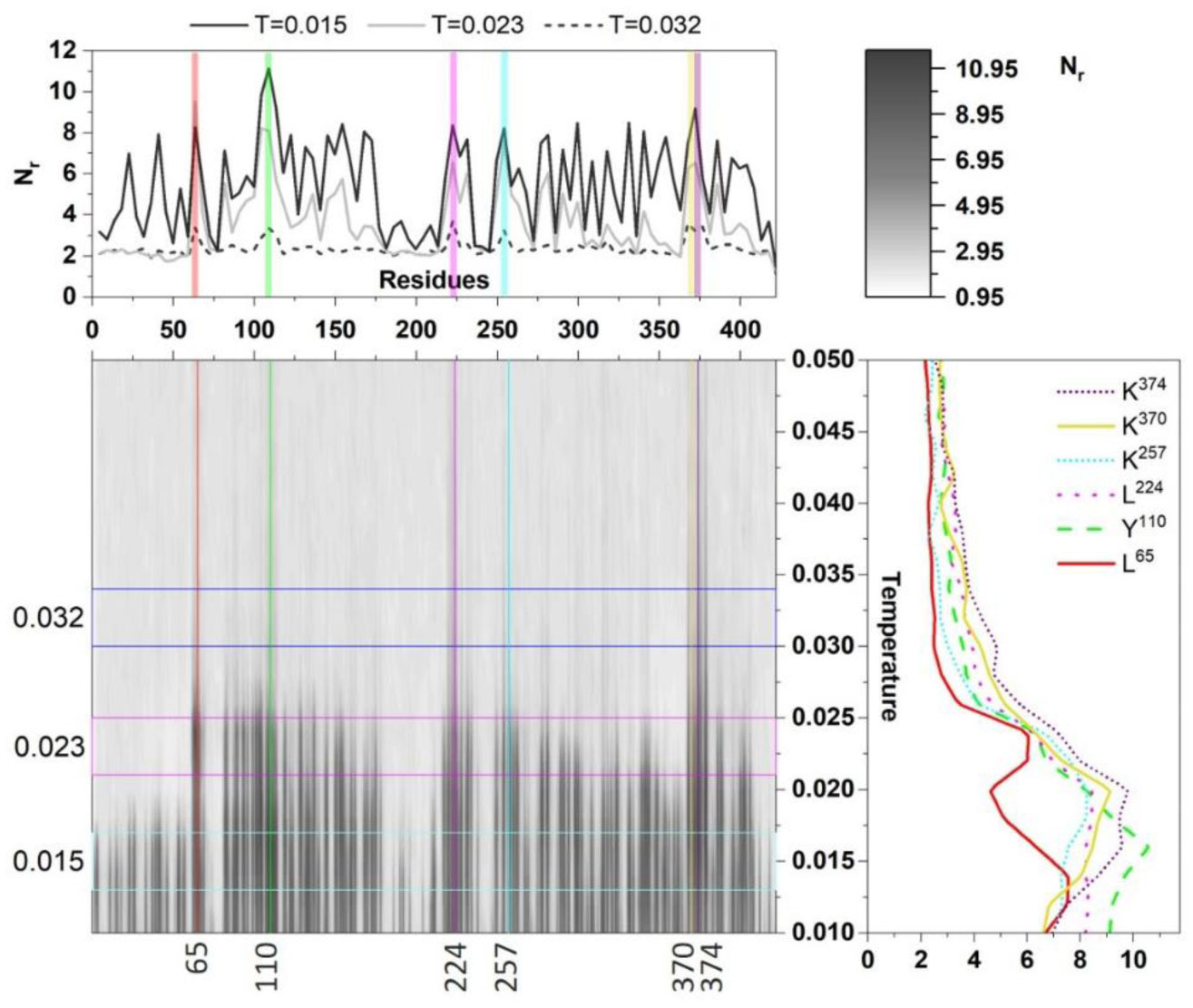
Figures(3)

Warin Rangubpit, Pornthep Sompornpisut, R.B. Pandey. Thermal-induced unfolding-refolding of a nucleocapsid COVN protein[J]. AIMS Biophysics, 2021, 8(1): 103-110. doi: 10.3934/biophy.2021007







 DownLoad:
DownLoad: