Citation: Moncef ATI, Rachid BOUAMRANE, Djamel ADDI, Fatima Zohra MAROC, Fatima Zohra Mecheret. Monte Carlo dose index estimation in computed tomography[J]. AIMS Bioengineering, 2020, 7(4): 224-235. doi: 10.3934/bioeng.2020019

| [1] | Ghavami SM, Mesbahi A, Pesianian I (2012) Patient doses from X-ray computed tomography examinations by a single array detector unit: Axial versus spiral mode. Int J Radiat Res 10: 89-94. |
| [2] |
Ngaile JE, Msaki PK (2006) Estimation of patient organ doses from CT examinations in Tanzania. J Appl Clin Med Phys 7: 80-94. doi: 10.1120/jacmp.v7i3.2200

|
| [3] | Saravanakumar A, Vaideki K, Govindarajan KN, et al. (2016) Establishment of CT diagnostic reference levels in select procedures in South India. Int J Radiat Res 14: 341-347. |
| [4] |
Kramer R, Cassola VF, Andrade MEA, et al. (2017) Mathematical modelling of scanner-specific bowtie filters for Monte Carlo CT dosimetry. Phys Med Biol 62: 781-809. doi: 10.1088/1361-6560/aa5343
|
| [5] | (2006) National Research CouncilHealth Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII Phase 2. Washington DC: The National Academies Press. |
| [6] | ICRP, ICRP Publication 103, The 2007 Recommendations of the International Commission on Radiological Protection, 2007.Available from: https://journals.sagepub.com/doi/pdf/10.1177/ANIB_37_2-4. |
| [7] |
Liu FL, Wang G, Cong WX, et al. (2013) Dynamic bowtie for fan-beam CT. J X-Ray Sci Technol 21: 579-590. doi: 10.3233/XST-130386
|
| [8] | Williams LE (2003) Therapeutic applications of Monte Carlo calculations in nuclear medicine. J Nucl Med 44: 991. |
| [9] | Fallah Mohammadi GR, Riyahi Alam N, Geraily G, et al. (2016) Thorax organ dose estimation in computed tomography based on patient CT data using Monte Carlo simulation. Int J Radiat Res 14: 313-321. |
| [10] |
Guidez J, Saturnin A (2017) Evolution of the collective radiation dose of nuclear reactors from the 2nd through to the 3rd generation and 4th generation sodium-cooled fast reactors. EPJ Nuclear Sci Technol 3: 32. doi: 10.1051/epjn/2017024
|
| [11] |
Vafapour H, Salehi Z (2018) Comparison of incident air kerma (ki) of common digital and analog radiology procedures in kohgiluyeh and boyer-Ahmad province. Pol J Med Phys Eng 24: 37-41. doi: 10.2478/pjmpe-2018-0006
|
| [12] | Cranley K, Gilmore BJ, Fogarty GWA (1991) Data for Estimating X-ray Tube Total Filtration UK: Institute of Physical Sciences in Medicine. |
| [13] |
Sahbaee P, Segars W P, Samei E (2014) Patient-based estimation of organ dose for a population of 58 adult patients across 13 protocol categories. Med Phys 41: 072104. doi: 10.1118/1.4883778
|
| [14] |
Birch R, Marshall M (1979) Computation of bremsstrahlung X-ray spectra and comparison with spectra measured with a Ge (Li) detector. Phys Med Biol 24: 505-517. doi: 10.1088/0031-9155/24/3/002
|
| [15] |
Lee CL, Kim HJ, Chung YH, et al. (2009) GATE simulations of CTDI for CT dose. J Korean Phys Soc 54: 1702-1708. doi: 10.3938/jkps.54.1702
|
| [16] | Poiata A, Focea R, Creanga D (2012) Pathogen germs response to low-dose radiation—medical approach. EDP Sci 24: 06005. |
| [17] | (2012) Comparison of different methods for measuring CT dose profiles with a new dosimetry phantom. Available form: http://www.irpa.net/members/P02.156.pdf. |
| [18] |
Judy PF, Balter S, Bassano D, et al. (1977) Phantoms for performance evaluation and quality assurance of CT scanners USA: American Association of Physicists in Medicine. doi: 10.37206/1
|
| [19] | Panzer W, Shrimpton PC, Jessen K, et al.European guidelines on quality criteria for computed tomography, EU publications, 2000.Available form: https://op.europa.eu/en/publication-detail/-/publication/d229c9e1-a967-49de-b169-59ee68605f1a. |
| [20] |
Siewerdsen JH, Waese AM, Moseley DJ, et al. (2004) Spektr: A computational tool for X-ray spectral analysis and imaging system optimization. Med Phys 31: 3057-3067. doi: 10.1118/1.1758350
|
| [21] | Nagel HD (2000) Radiation Exposure in Computed Tomography: Fundamentals, Influencing Parameters, Dose Assessment, Optimisation, Scanner Data, Terminology Hamburg: Offizin Paul Hartung Druck GmbH & Co. KGD-20020. |
| [22] |
Hernandez AM, Boone JM (2014) Tungsten anode spectral model using interpolating cubic splines: unfiltered X-ray spectra from 20 kV to 640 kV. Med Phys 41: 042101. doi: 10.1118/1.4866216
|
| [23] |
Khoramian D, Sistani S, Hejazi P (2019) Establishment of diagnostic reference levels arising from common CT examinations in Semnan County, Iran. Pol J Med Phys Eng 25: 51-55. doi: 10.2478/pjmpe-2019-0008
|
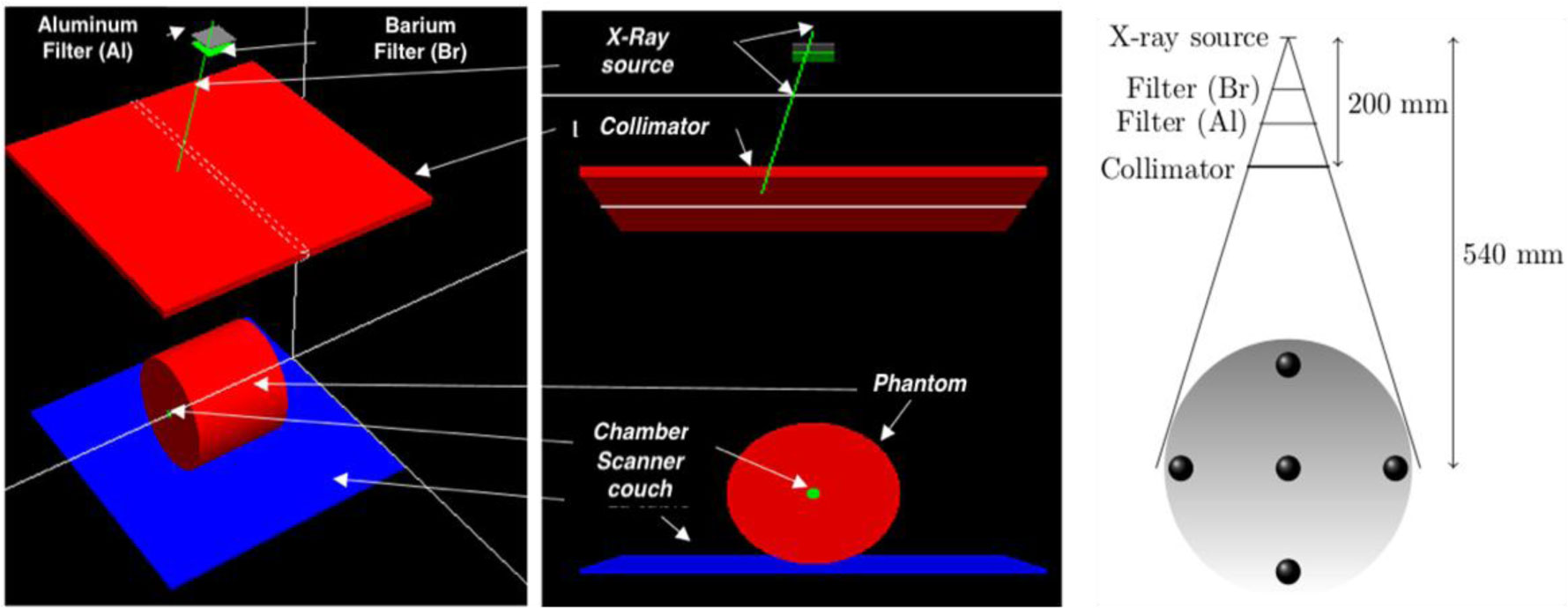
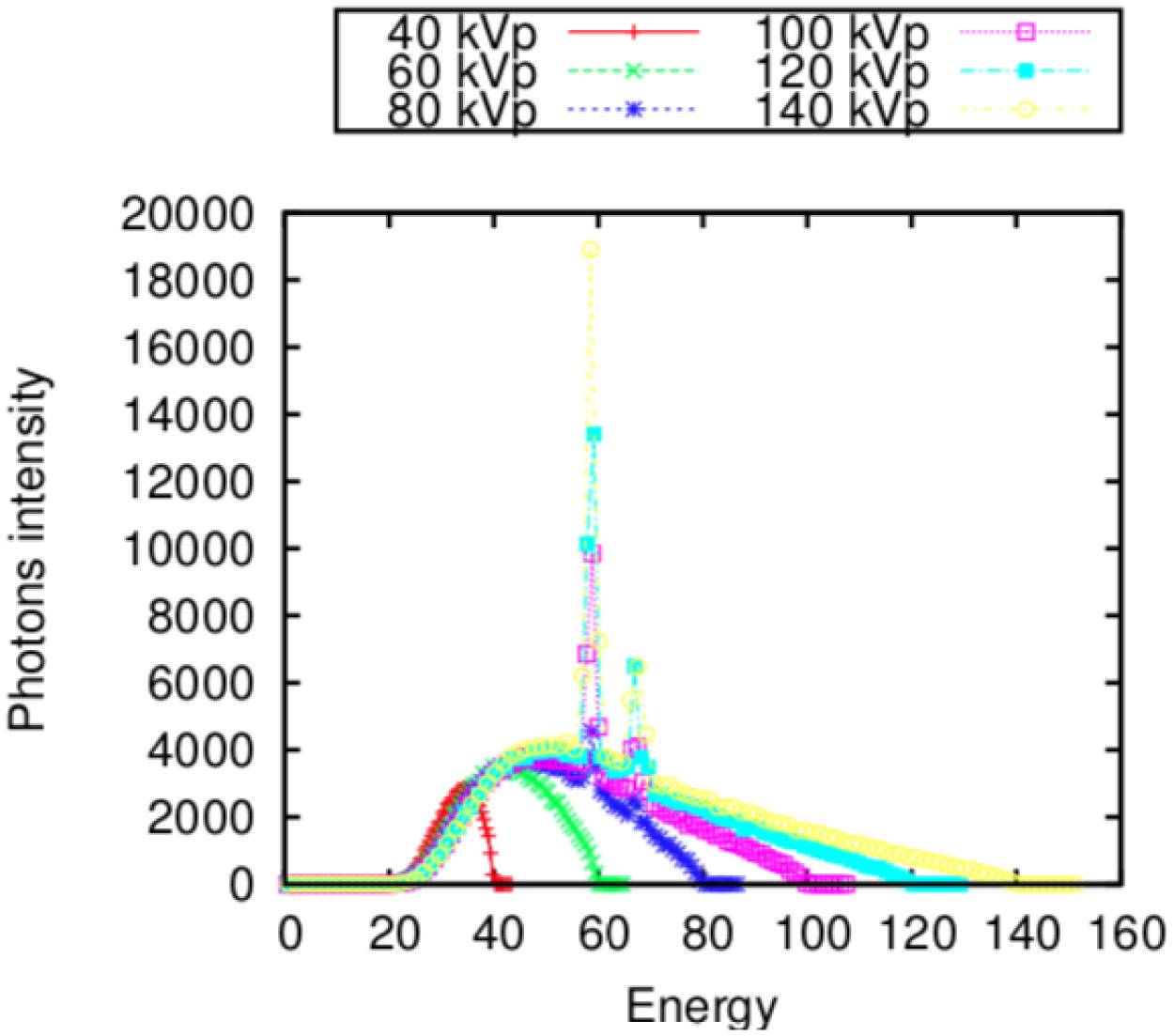
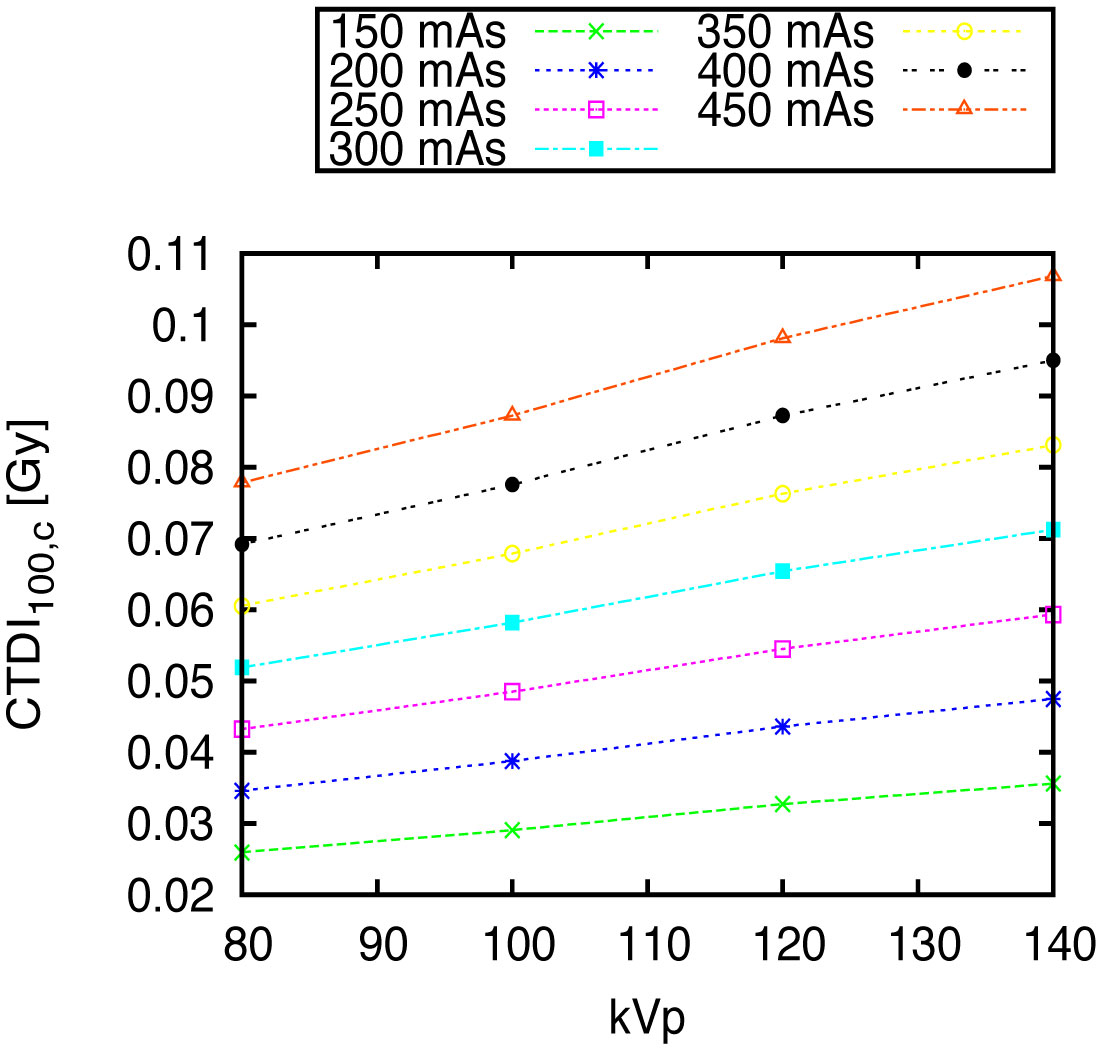
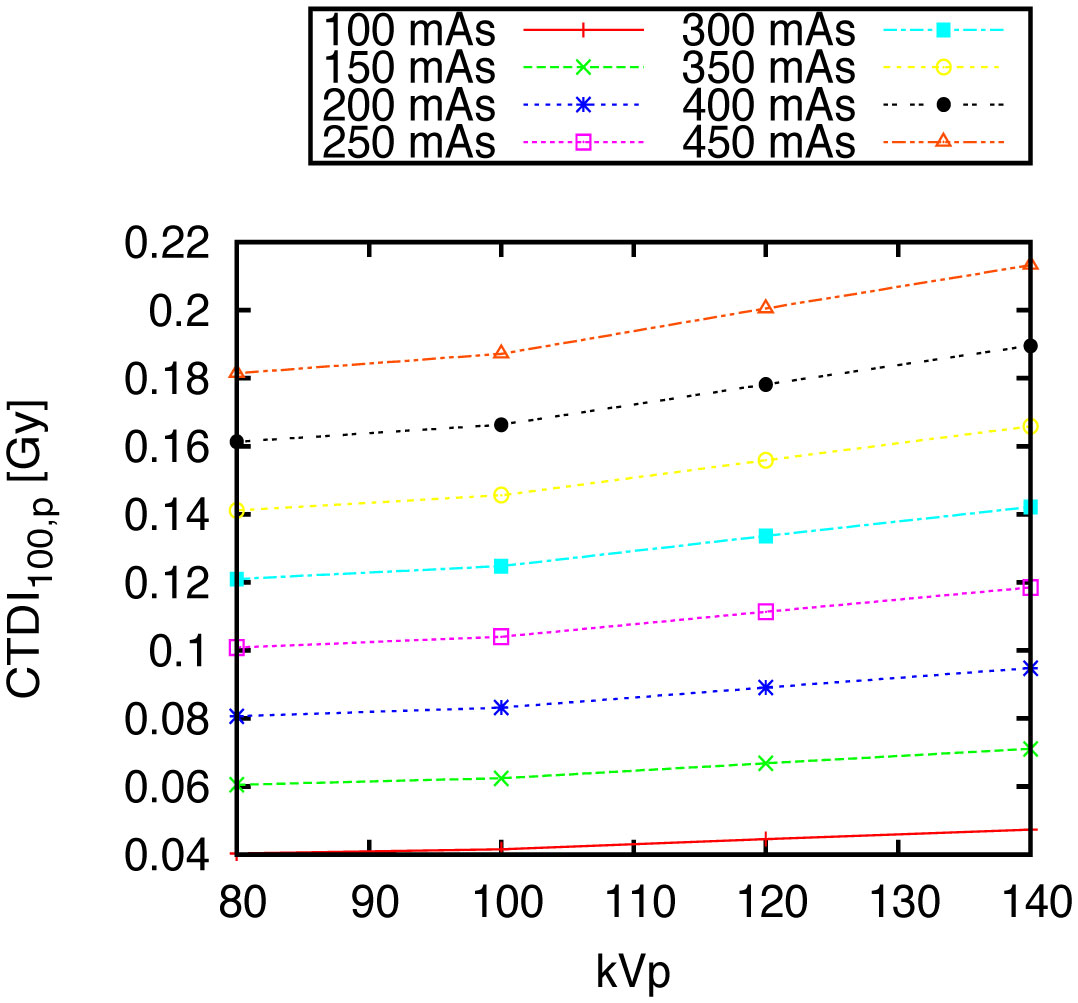
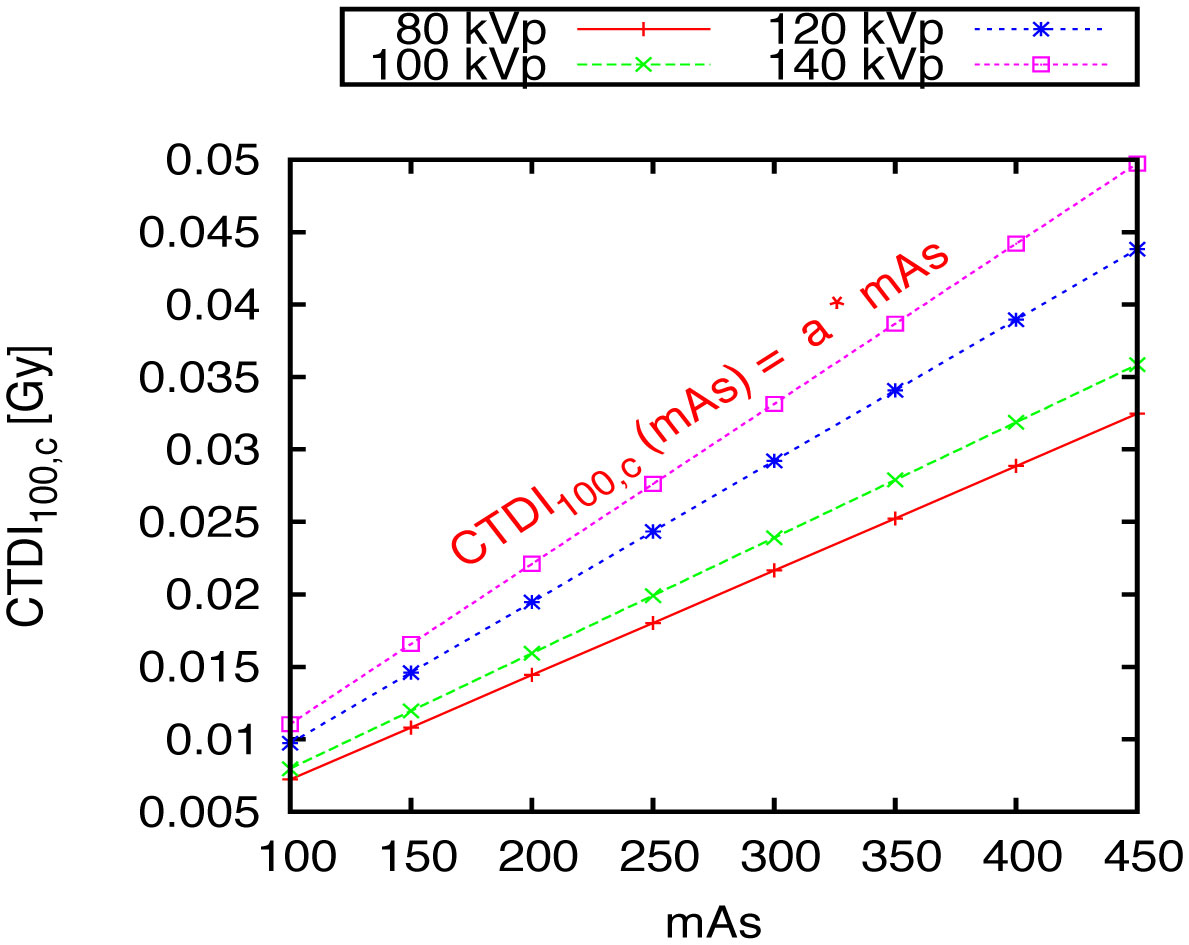
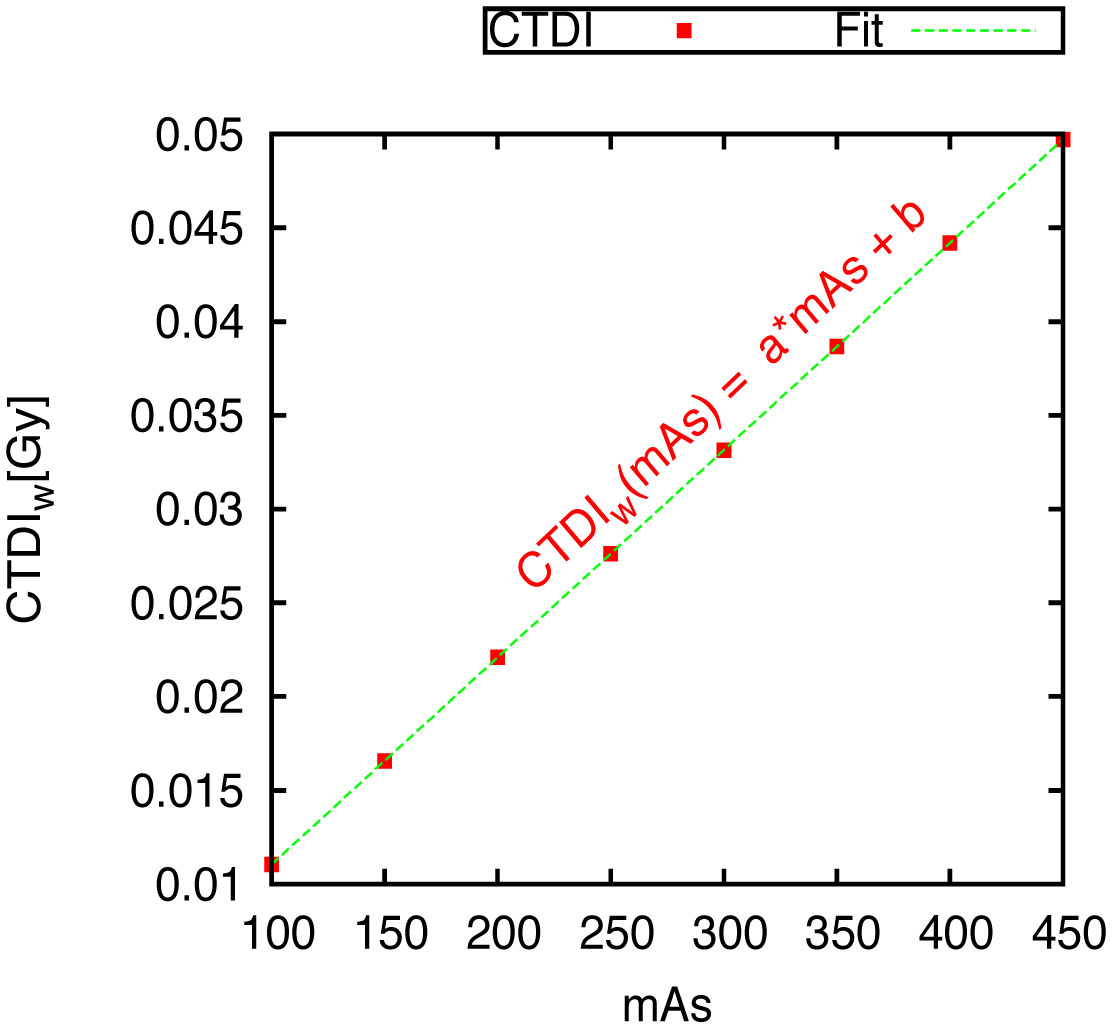
Figures(10) / Tables(1)

Moncef ATI, Rachid BOUAMRANE, Djamel ADDI, Fatima Zohra MAROC, Fatima Zohra Mecheret. Monte Carlo dose index estimation in computed tomography[J]. AIMS Bioengineering, 2020, 7(4): 224-235. doi: 10.3934/bioeng.2020019






 DownLoad:
DownLoad: