Citation: Jian Zou, Fulton T. Crews. Glutamate/NMDA excitotoxicity and HMGB1/TLR4 neuroimmune toxicity converge as components of neurodegeneration[J]. AIMS Molecular Science, 2015, 2(2): 77-100. doi: 10.3934/molsci.2015.2.77

| [1] |
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1: 120-129. doi: 10.1038/35040009

|
| [2] |
Bredesen DE, Rao RV, Mehlen P (2006) Cell death in the nervous system. Nature 443: 796-802. doi: 10.1038/nature05293
|
| [3] |
Qin L, Wu X, Block ML, et al. (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: 453-462. doi: 10.1002/glia.20467
|
| [4] |
Tilleux S, Hermans E (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85: 2059-2070. doi: 10.1002/jnr.21325
|
| [5] |
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51: 333-355. doi: 10.1016/j.neuint.2007.03.012
|
| [6] | Kostic M, Zivkovic N, Stojanovic I (2013) Multiple sclerosis and glutamate excitotoxicity. Rev Neurosci 24: 71-88. |
| [7] |
Morimoto K, Murasugi T, Oda T (2002) Acute neuroinflammation exacerbates excitotoxicity in rat hippocampus in vivo. Exp Neurol 177: 95-104. doi: 10.1006/exnr.2002.7991
|
| [8] |
Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 1034: 11-24. doi: 10.1016/j.brainres.2004.11.014
|
| [9] |
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191-195. doi: 10.1038/nature00858
|
| [10] |
Bell CW, Jiang W, Reich CF, et al. (2006) The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291: C1318-1325. doi: 10.1152/ajpcell.00616.2005
|
| [11] |
Zou JY, Crews FT (2014) Release of neuronal HMGB1 by Ethanol through decreased HDAC activity activates brain neuroimmune signaling. PloS One 9: e87915. doi: 10.1371/journal.pone.0087915
|
| [12] |
Evankovich J, Cho SW, Zhang R, et al. (2010) High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem 285: 39888-39897. doi: 10.1074/jbc.M110.128348
|
| [13] |
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331-342. doi: 10.1038/nri1594
|
| [14] |
Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29: 139-162. doi: 10.1146/annurev-immunol-030409-101323
|
| [15] | Tang D, Billiar TR, Lotze MT (2012) A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med 18: 1360-1362. |
| [16] |
Janko C, Filipovic M, Munoz LE, et al. (2014) Redox modulation of HMGB1-related signaling. Antioxid Redox Signal 20: 1075-1085. doi: 10.1089/ars.2013.5179
|
| [17] | Magna M, Pisetsky DS (2014) The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med 20: 138-146. |
| [18] |
Faraco G, Fossati S, Bianchi ME, et al. (2007) High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem 103: 590-603. doi: 10.1111/j.1471-4159.2007.04788.x
|
| [19] |
Kim JB, Sig Choi J, Yu YM, et al. (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26: 6413-6421. doi: 10.1523/JNEUROSCI.3815-05.2006
|
| [20] |
Lo Coco D, Veglianese P, Allievi E, et al. (2007) Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci Lett 412: 73-77. doi: 10.1016/j.neulet.2006.10.063
|
| [21] | Crews FT, Qin L, Sheedy D, et al. (2012) High Mobility Group Box 1/Toll-like Receptor Danger Signaling Increases Brain Neuroimmune Activation in Alcohol Dependence. Biol Psychiatry 73: 602-612. |
| [22] |
Maroso M, Balosso S, Ravizza T, et al. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 16: 413-419. doi: 10.1038/nm.2127
|
| [23] |
Kim SW, Jin Y, Shin JH, et al. (2012) Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti-inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol Dis 46: 147-156. doi: 10.1016/j.nbd.2011.12.056
|
| [24] |
Ohnishi M, Katsuki H, Fukutomi C, et al. (2011) HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology 61: 975-980. doi: 10.1016/j.neuropharm.2011.06.026
|
| [25] |
Okuma Y, Liu K, Wake H, et al. (2014) Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 85: 18-26. doi: 10.1016/j.neuropharm.2014.05.007
|
| [26] | Chavan SS, Huerta PT, Robbiati S, et al. (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Molecular medicine 18: 930-937. |
| [27] |
Brana C, Benham C, Sundstrom L (2002) A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc 10: 109-114. doi: 10.1016/S1385-299X(02)00201-5
|
| [28] |
Noraberg J, Kristensen BW, Zimmer J (1999) Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc 3: 278-290. doi: 10.1016/S1385-299X(98)00050-6
|
| [29] |
Zimmer J, Kristensen BW, Jakobsen B, et al. (2000) Excitatory amino acid neurotoxicity and modulation of glutamate receptor expression in organotypic brain slice cultures. Amino Acids 19: 7-21. doi: 10.1007/s007260070029
|
| [30] |
Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20: 83-90. doi: 10.1016/0165-0270(87)90041-0
|
| [31] |
Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 1034: 11-24. doi: 10.1016/j.brainres.2004.11.014
|
| [32] |
Balosso S, Liu J, Bianchi ME, et al. (2014) Disulfide-Containing High Mobility Group Box-1 Promotes N-Methyl-d-Aspartate Receptor Function and Excitotoxicity by Activating Toll-Like Receptor 4-Dependent Signaling in Hippocampal Neurons. Antioxid Redox Signal 21: 1726-1740. doi: 10.1089/ars.2013.5349
|
| [33] | Chenard BL, Menniti FS (1999) Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharma Des 5: 381-404. |
| [34] |
Kim SW, Lim CM, Kim JB, et al. (2011) Extracellular HMGB1 released by NMDA treatment confers neuronal apoptosis via RAGE-p38 MAPK/ERK signaling pathway. Neurotox Res 20: 159-169. doi: 10.1007/s12640-010-9231-x
|
| [35] |
Glass CK, Saijo K, Winner B, et al. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918-934. doi: 10.1016/j.cell.2010.02.016
|
| [36] |
Crews FT, Qin L, Sheedy D, et al. (2013) High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 73: 602-612. doi: 10.1016/j.biopsych.2012.09.030
|
| [37] |
Kim JB, Lim CM, Yu YM, et al. (2008) Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res 86: 1125-1131. doi: 10.1002/jnr.21555
|
| [38] |
Qiu J, Nishimura M, Wang Y, et al. (2008) Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab 28: 927-938. doi: 10.1038/sj.jcbfm.9600582
|
| [39] | Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7: 369-379. |
| [40] |
Kim ID, Shin JH, Lee HK, et al. (2012) Intranasal delivery of HMGB1-binding heptamer peptide confers a robust neuroprotection in the postischemic brain. Neurosci Lett 525: 179-183. doi: 10.1016/j.neulet.2012.07.040
|
| [41] |
Sun Q, Wang F, Li W, et al. (2013) Glycyrrhizic acid confers neuroprotection after subarachnoid hemorrhage via inhibition of high mobility group box-1 protein: a hypothesis for novel therapy of subarachnoid hemorrhage. Med Hypotheses 81: 681-685. doi: 10.1016/j.mehy.2013.07.026
|
| [42] |
Zhang J, Wu Y, Weng Z, et al. (2014) Glycyrrhizin protects brain against ischemia-reperfusion injury in mice through HMGB1-TLR4-IL-17A signaling pathway. Brain Res 1582: 176-186. doi: 10.1016/j.brainres.2014.07.002
|
| [43] | Yang QW, Xiang J, Zhou Y, et al. (2010) Targeting HMGB1/TLR4 signaling as a novel approach to treatment of cerebral ischemia. Front Biosci 2: 1081-1091. |
| [44] |
Laird MD, Shields JS, Sukumari-Ramesh S, et al. (2014) High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 62: 26-38. doi: 10.1002/glia.22581
|
| [45] |
Maroso M, Balosso S, Ravizza T, et al. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 16: 413-419. doi: 10.1038/nm.2127
|
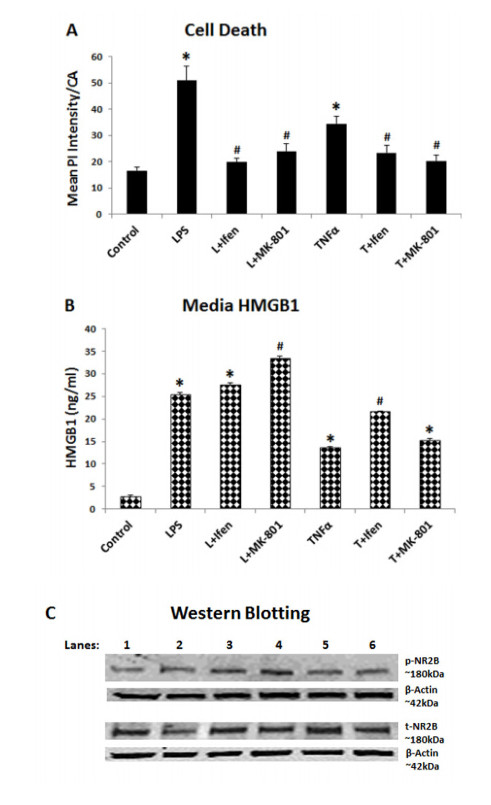
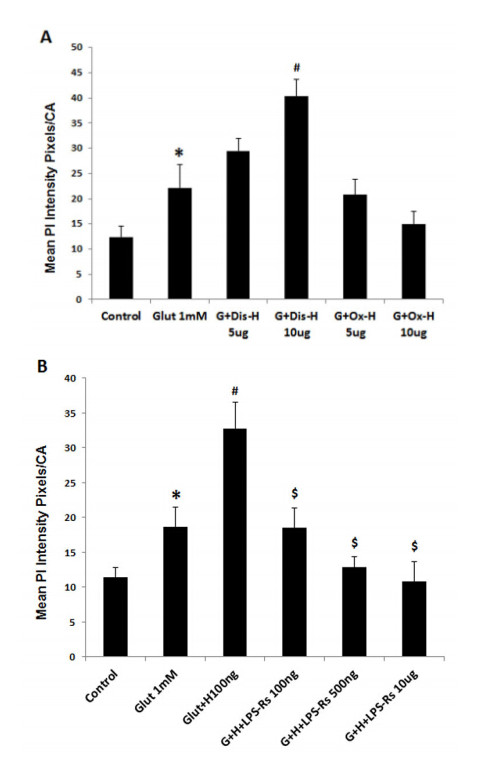
Figures(10)

Jian Zou, Fulton T. Crews. Glutamate/NMDA excitotoxicity and HMGB1/TLR4 neuroimmune toxicity converge as components of neurodegeneration[J]. AIMS Molecular Science, 2015, 2(2): 77-100. doi: 10.3934/molsci.2015.2.77







 DownLoad:
DownLoad: