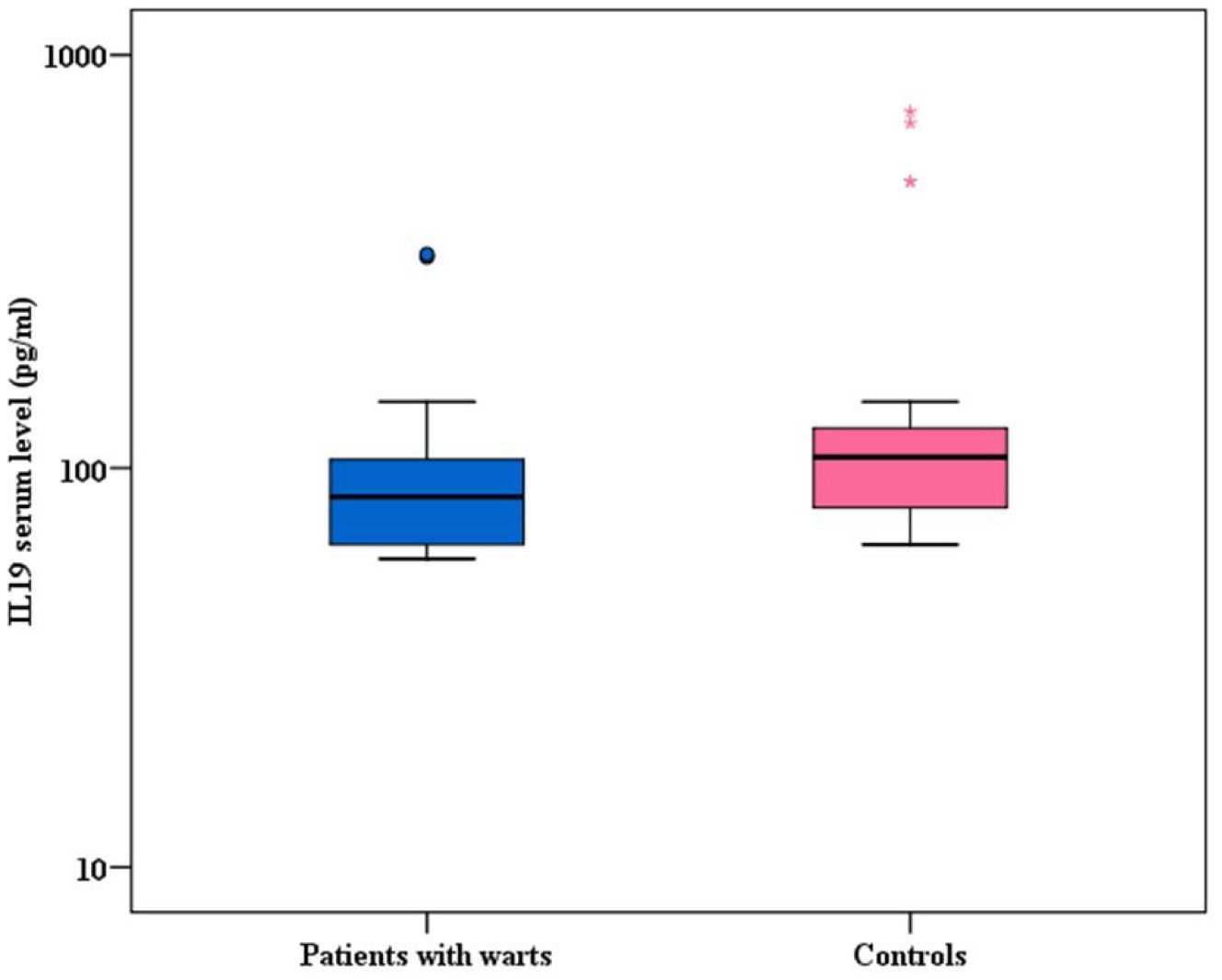
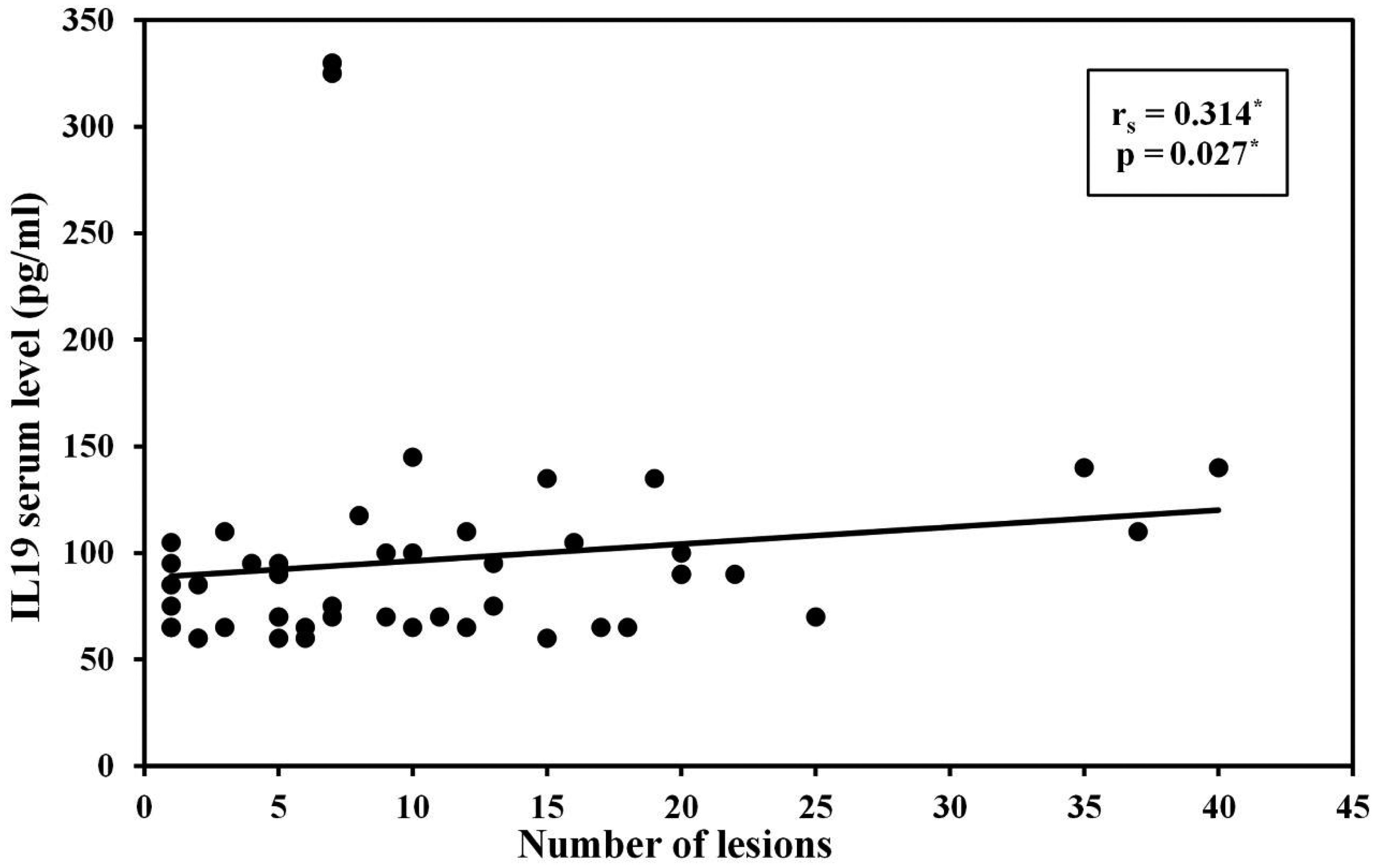
Background: Warts are viral cutaneous infections caused by human papilloma virus (HPV), presented by verrucous growth over the skin surface. The cell mediated immune response is considered to play a crucial role in HPV clearance. The viral load and number of lesions increase when there is an imbalance between the T-helper 1 and T-helper 2 immune responses. Interleukin (IL)-19 is a cytokine that belongs to interleukin 10 cytokines family and constitutes a sub-family with IL-20, IL-22 and IL-24. IL-19 is mainly produced by activated monocytes and to a lesser extent by B-cells, keratinocytes and fetal membranes. IL-19 was found to shift T-cell maturation away from the pro-inflammatory T-helper 1 cells toward the anti-inflammatory T-helper 2 cells. It induces IL-4 and IL-13 production in T cells and apoptosis in monocytes. Aim: This study aimed to measure serum level of IL-19 in patients with warts compared to healthy controls and to find out the correlation between this level and number, size and clinical types of warts. Methods: The study included 50 patients with warts and 50 control subjects. Serum concentration of IL-19 was measured by enzyme-linked immune sorbent assay. Results: Interleukin-19 serum level was significantly lower in patients with warts than in controls (P < 0.003). Moreover, there was a significant positive correlation between IL-19 serum level and the number of warts (P = 0.027). Conclusion: Serum level of IL-19 was significantly lower in patients with warts, and this low level might be crucial for an effective cell mediated immunological response to HPV.
Citation: Radwa El-Sayed Mahmoud Marie, Aya Mohamed kamal Lasheen, Hebat-Allah Hassan Nashaat, Mona A. Atwa. Assessment of serum interleukin 19 level in patients with warts[J]. AIMS Molecular Science, 2023, 10(1): 1-10. doi: 10.3934/molsci.2023001
Background: Warts are viral cutaneous infections caused by human papilloma virus (HPV), presented by verrucous growth over the skin surface. The cell mediated immune response is considered to play a crucial role in HPV clearance. The viral load and number of lesions increase when there is an imbalance between the T-helper 1 and T-helper 2 immune responses. Interleukin (IL)-19 is a cytokine that belongs to interleukin 10 cytokines family and constitutes a sub-family with IL-20, IL-22 and IL-24. IL-19 is mainly produced by activated monocytes and to a lesser extent by B-cells, keratinocytes and fetal membranes. IL-19 was found to shift T-cell maturation away from the pro-inflammatory T-helper 1 cells toward the anti-inflammatory T-helper 2 cells. It induces IL-4 and IL-13 production in T cells and apoptosis in monocytes. Aim: This study aimed to measure serum level of IL-19 in patients with warts compared to healthy controls and to find out the correlation between this level and number, size and clinical types of warts. Methods: The study included 50 patients with warts and 50 control subjects. Serum concentration of IL-19 was measured by enzyme-linked immune sorbent assay. Results: Interleukin-19 serum level was significantly lower in patients with warts than in controls (P < 0.003). Moreover, there was a significant positive correlation between IL-19 serum level and the number of warts (P = 0.027). Conclusion: Serum level of IL-19 was significantly lower in patients with warts, and this low level might be crucial for an effective cell mediated immunological response to HPV.
Interleukin
Human papilloma virus
T-helper
Enzyme-Linked Immune-sorbent Assay
Horseradish Peroxidase
optical density
Hepatitis B virus

| [1] |
Paaso A, Koskimaa HM, Welters MJ, et al. (2015) Cell mediated immunity against HPV16 E2, E6 and E7 peptides in women with incident CIN and in constantly HPV-negative women followed-up for 10-years. J Transl Med 13: 1-11. https://doi.org/10.1186/s12967-015-0498-9 
|
| [2] |
Steinbach A, Riemer AB (2018) Immune evasion mechanisms of human papillomavirus. Int J Cancer 142: 224-229. https://doi.org/10.1002/ijc.31027
|
| [3] |
Fujimoto Y, Azuma YT, Matsuo Y, et al. (2017) Interleukin-19 contributes as a protective factor in experimental Th2-mediated colitis. Naunyn. Schmiedebergs. Arch Pharmacol 390: 261-268. https://doi.org/10.1007/s00210-016-1329-0
|
| [4] |
Sabat R (2010) IL-10 family of cytokines. Cytokine Growth Factor Rev 21: 315-324. https://doi.org/10.1016/j.cytogfr.2010.11.001
|
| [5] |
Gallagher G, Eskdale J, Jordan W, et al. (2004) Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. Int Immunopharmacol 4: 615-626. https://doi.org/10.1016/j.intimp.2004.01.005
|
| [6] |
Schiffman M, Doorbar J, Wentzensen N, et al. (2016) Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2: 16086. https://doi.org/10.1038/nrdp.2016.86
|
| [7] |
Fernandes JV, DE Medeiros Fernandes TA, DE Azevedo JC, et al. (2015) Link between chronic inflammation and human papillomavirus-induced carcinogenesis (Review). Oncol Lett 9: 1015-1026. https://doi.org/10.3892/ol.2015.2884
|
| [8] |
Liao SC, Cheng YC, Wang YC, et al. (2004) IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol 173: 6712-6718. https://doi.org/10.4049/jimmunol.173.11.6712
|
| [9] |
Oguejiofor K, Hall J, Slater C, et al. (2015) Stromal infiltration of CD8 T cells is associated with improved clinical outcome in HPV-positive oropharyngeal squamous carcinoma. Br J Cancer 113: 886-893. https://doi.org/10.1038/bjc.2015.277
|
| [10] |
Azuma YT, Nishiyama K (2020) Interleukin-19 enhances cytokine production induced by lipopolysaccharide and inhibits cytokine production induced by polyI:C in BALB/c mice. J Vet Med Sc 82: 891-896. https://doi.org/10.1292/jvms.20-0137
|
| [11] |
Liao YC, Liang WG, Chen FW, et al. (2002) IL-19 induces production of IL-6 and TNF-alpha and results in cell apoptosis through TNF-alpha. J Immunol 169: 4288-4297. https://doi.org/10.4049/jimmunol.169.8.4288
|
| [12] |
Hsu YH, Li HH, Sung JM, et al. (2013) Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury. PLoS One 8: e56028. https://doi.org/10.1371/journal.pone.0056028
|
| [13] |
AH, Esawy AM, Khalifa NA, et al. (2020) Evaluation of serum interleukin 17 and zinc levels in recalcitrant viral wart. J Cosmet Dermatol 19: 954-959. https://doi.org/10.1111/jocd.13106
|
| [14] |
Xu X, Prens E, Florencia E, et al. (2021) Interleukin-17A Drives IL-19 and IL-24 Expression in Skin Stromal Cells Regulating Keratinocyte Proliferation. Front Immunol 12: 719562. https://doi.org/10.3389/fimmu.2021.719562
|
| [15] |
Wang Y, Liu X, Li Y, et al. (2013) The paradox of IL-10-mediated modulation in cervical cancer (Review). Biomedical Reports 1: 347-351. https://doi.org/10.3892/br.2013.69
|
| [16] |
Berti FCB, Pereira APL, Cebinelli GCM, et al. (2017) The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor Rev 34: 1-13. https://doi.org/10.1016/j.cytogfr.2017.03.002
|
| [17] |
Marie REM, Abuzeid AQEM, Attia FM, et al. (2021) Serum level of interleukin-22 in patients with cutaneous warts: A case-control study. J Cosmet Dermatol 2021: 1782-1787. https://doi.org/10.1111/jocd.13779
|
Figures(2) / Tables(5)

Radwa El-Sayed Mahmoud Marie, Aya Mohamed kamal Lasheen, Hebat-Allah Hassan Nashaat, Mona A. Atwa. Assessment of serum interleukin 19 level in patients with warts[J]. AIMS Molecular Science, 2023, 10(1): 1-10. doi: 10.3934/molsci.2023001







 DownLoad:
DownLoad: