Citation: Nicole Shamitko-Klingensmith, Jonathan W. Boyd, Justin Legleiter. Microtubule modification influences cellular response to amyloid-β exposure[J]. AIMS Biophysics, 2016, 3(2): 261-285. doi: 10.3934/biophy.2016.2.261

| [1] |
Prokop A, Beaven R, Qu Y, et al. (2013) Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J Cell Sci 126: 2331–2341. doi: 10.1242/jcs.126912

|
| [2] |
Iqbal K, Zaidi T, Wen G, et al. (1986) Defective brain microtubule assembly in Alzheimer's disease. The Lancet 328: 421–426. doi: 10.1016/S0140-6736(86)92134-3
|
| [3] |
Hirokawa N, Takeda S (1998) Gene targeting studies begin to reveal the function of neurofilament proteins. J Cell Biol 143: 1143–1143. doi: 10.1083/jcb.143.4.1143
|
| [4] |
Riederer IM, Robert P, Porchet R, et al. (2003) Selective changes in the neurofilament and microtubule cytoskeleton of NF-H/LacZ mice. J Neurosci Res 71: 196–207. doi: 10.1002/jnr.10485
|
| [5] |
Twelvetrees A, Hendricks AG, Holzbaur ELF (2012) SnapShot: Axonal Transport. Cell 149: 950–U251. doi: 10.1016/j.cell.2012.05.001
|
| [6] |
Elder GA, Friedrich VL, Margita A, et al. (1999) Age-related atrophy of motor axons in mice deficient in the mid-sized neurofilament subunit. J Cell Biol 146: 181–192. doi: 10.1083/jcb.146.1.181
|
| [7] |
Gourlay CW, Ayscough KR (2005) The actin cytoskeleton in ageing and apoptosis. FEMS Yeast Res 5: 1193–1198. doi: 10.1016/j.femsyr.2005.08.001
|
| [8] |
Gourlay CW, Carpp LN, Timpson P, et al. (2004) A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol 164: 803–809. doi: 10.1083/jcb.200310148
|
| [9] |
Parhad IM, Scott JN, Cellars LA, et al. (1995) Axonal atrophy in aging is associated with a decline in neurofilament gene expression. J Neurosci Res 41: 355–366. doi: 10.1002/jnr.490410308
|
| [10] |
Raes M (1991) Involvement of microtubules in modifications associated with cellular aging. Mutat Res 256: 149–168. doi: 10.1016/0921-8734(91)90008-Y
|
| [11] | Uchida A, Yorifuji H, Lee VMY, et al. (1999) Neurofilaments of aged rats: The strengthened interneurofilament interaction and the reduced amount of NF-M. J Neurosci Res 58: 337–348. |
| [12] |
Vickers JC, Riederer BM, Marugg RA, et al. (1994) Alterations in neurofilament protein immunoreactivity in human hippocampal neurons related to normal aging and Alzheimers disease. Neuroscience 62: 1–13. doi: 10.1016/0306-4522(94)90310-7
|
| [13] |
Bruusgaard JC, Liestøl K, Gundersen K (2006) Distribution of myonuclei and microtubules in live muscle fibers of young, middle-aged, and old mice. J Appl Physiol 100:2024–2030. doi: 10.1152/japplphysiol.00913.2005
|
| [14] |
Raes M, Geuens G, de Brabander M, et al. (1983) Microtubules and microfilaments in ageing hamster embryo fibroblasts in vitro. Exp Gerontol 18: 241–254. doi: 10.1016/0531-5565(83)90035-9
|
| [15] |
Raes M, Remacle J (1987) Alteration of the microtubule organization in aging WI-38 fibroblasts - a comparative study with embryonic hamster lung fibroblasts. Exp Gerontol 22: 47–58. doi: 10.1016/0531-5565(87)90014-3
|
| [16] |
Saha S, Slepecky NB (2000) Age-related changes in microtubules in the guinea pig organ of Corti - Tubulin isoform shifts with increasing age suggest changes in micromechanical properties of the sensory epithelium. Cell Tissue Res 300: 29–46. doi: 10.1007/s004410050045
|
| [17] | Neely MD, Sidell KR, Graham DG, et al. (1999) The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J Neurochem 72: 2323–2333. |
| [18] |
Cash AD, Aliev G, Siedlak SL, et al. (2003) Microtubule reduction in Alzheimer's disease and aging is independent of τ filament formation. Am J Pathol 162: 1623–1627. doi: 10.1016/S0002-9440(10)64296-4
|
| [19] |
Davies DC, Horwood N, Isaacs SL, et al. (1992) The effect of age and Alzheimer's disease on pyramidal neuron density in the individual fields of the hippocampal formation. Acta Neuropathol 83: 510–517. doi: 10.1007/BF00310028
|
| [20] | Karbowski M, Spodnik JH, Teranishi M, et al. (2001) Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J Cell Sci 114: 281–291. |
| [21] |
Praprotnik D, Smith MA, Richey PL, et al. (1996) Filament heterogeneity within the dystrophic neurites of senile plaques suggests blockage of fast axonal transport in Alzheimer's disease. Acta Neuropathol 91: 226–235. doi: 10.1007/s004010050420
|
| [22] |
Terry RD, Katzman R (2001) Life span and synapses: will there be a primary senile dementia? Neurobiol Aging 22: 347–348. doi: 10.1016/S0197-4580(00)00250-5
|
| [23] |
Mattson MP, Magnus T (2006) Aging and neuronal vulnerability. Nat Rev Neurosci 7: 278–294. doi: 10.1038/nrn1886
|
| [24] | Selkoe DJ (2011) Alzheimer’s Disease. Cold Spring Harb Perspect Biol 3: a004457. |
| [25] |
Zempel H, Thies E, Mandelkow E, et al. (2010) Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30: 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
|
| [26] |
De Felice FG, Wu D, Lambert MP, et al. (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol Aging 29: 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
|
| [27] | Jin M, Shepardson N, Yang T, et al. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. P Natl Acad Sci USA 108: 5819–5824. |
| [28] |
Busciglio J, Lorenzo A, Yeh J, et al. (1995) β-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14: 879–888. doi: 10.1016/0896-6273(95)90232-5
|
| [29] |
Ekinci FJ, Malik KU, Shea TB (1999) Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to β-amyloid - Map kinase mediates β-amyloid-induced neurodegeneration. J Biol Chem 274: 30322–30327. doi: 10.1074/jbc.274.42.30322
|
| [30] |
Ferreira A, Lu Q, Orecchio L, et al. (1997) Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar Aβ. Mol Cell Neurosci 9: 220–234. doi: 10.1006/mcne.1997.0615
|
| [31] |
Lorenzo A, Yankner BA (1994) β-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. P Natl Acad Sci USA 91: 12243–12247. doi: 10.1073/pnas.91.25.12243
|
| [32] | Shea TB, Dergay AN, Ekinci FJ (1998) β-amyloid induced hyperphosphorylation of tau in human neuroblastoma cells involves map kinase. Neurosc Res Commun 22: 45–49. |
| [33] |
Takashima A, Noguchi K, Sato K, et al. (1993) Tau protein kinase is essential for amyloid β protein induced neurotoxicity. P Natl Acad Sci USA 90: 7789–7793. doi: 10.1073/pnas.90.16.7789
|
| [34] |
Cutler RG, Kelly J, Storie K, et al. (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. P Natl Acad Sci USA 101: 2070–2075. doi: 10.1073/pnas.0305799101
|
| [35] |
Nixon RA (2003) The calpains in aging and aging-related diseases. Ageing Res Rev 2: 407–418. doi: 10.1016/S1568-1637(03)00029-1
|
| [36] |
Stokin GB, Lillo C, Falzone TL, et al. (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307: 1282–1288. doi: 10.1126/science.1105681
|
| [37] |
Mellon PL, Windle JJ, Goldsmith PC, et al. (1990) Immortalization of hypothalamic GNRH neurons by genetically targeted tumorigenesis. Neuron 5: 1–10. doi: 10.1016/0896-6273(90)90028-E
|
| [38] |
Liposits Z, Merchenthaler I, Wetsel WC, et al. (1991) Morphological characterization of immortalized hypothalamic neurons synthesizing luteinizing hormone-releasing hormone. Endocrinology 129: 1575–1583. doi: 10.1210/endo-129-3-1575
|
| [39] |
Wetsel WC, Mellon PL, Weiner RI, et al. (1991) Metabolism of pro-luteinizing hormone releasing hormone in immortalized hypothalamic neurons. Endocrinology 129: 1584–1595. doi: 10.1210/endo-129-3-1584
|
| [40] |
Wetsel WC, Valenca MM, Merchenthaler I, et al. (1992) Intrinsic pulsatile secretory activity of immortalized luteinizing hormone releasing hormone secreting neurons. P Natl Acad Sci USA 89: 4149–4153. doi: 10.1073/pnas.89.9.4149
|
| [41] | Whyte DB, Lawson MA, Belsham DD, et al. (1995) A neuron specific enhancer targets expression of the gonadotropin releasing hormone gene to hypothalamic neurosecretory neurons. Mol Endocrinol 9: 467–477. |
| [42] |
Stine WB, Dahlgren KN, Krafft GA, et al. (2003) In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem 278: 11612–11622. doi: 10.1074/jbc.M210207200
|
| [43] | Rheinlaender J, Geisse NA, Proksch R, et al. (2010) Comparison of scanning ion conductance microscopy with atomic force microscopy for cell imaging. Langmuir 27: 697–704. |
| [44] |
Hutter JL, Bechhoefer J (1993) Calibration of atomic-force microscope tips. Rev Sci Instrum 64: 1868. doi: 10.1063/1.1143970
|
| [45] |
Burke KA, Godbey J, Legleiter J (2011) Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods 53: 275–284. doi: 10.1016/j.ymeth.2010.12.028
|
| [46] | Simakova O, Arispe NJ (2007) The cell-selective neurotoxicity of the Alzheimer's amyloid-β peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for amyloid-β toxicity. J Neurosci 27: 13719–13729. |
| [47] | Smiakova O, Arispe NJ (2011) Fluorescent Analysis of the Cell-Selective Alzheimer's Disease Amyloid-β Peptide Surface Membrane Binding: Influence of Membrane Components. Int J Alzheimer's Dis 2011: 917629. |
| [48] |
Alenghat FJ, Nauli SM, Kolb R, et al. (2004) Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp Cell Res 301: 23–30. doi: 10.1016/j.yexcr.2004.08.003
|
| [49] |
Rao S, Orr GA, Chaudhary AG, et al. (1995) Characterization of the taxol binding site on the microtubule: 2-(m-azidobenzoyl) taxol photolabels a peptide (amino acids 217-231) of β-tubulin. J Biol Chem 270: 20235–20238. doi: 10.1074/jbc.270.35.20235
|
| [50] |
Dennerll TJ, Joshi HC, Steel VL, et al. (1988) Tension and compression in the cytoskeleton of PC-12 neurites II - quantative measurements. J Cell Biol 107: 665–674. doi: 10.1083/jcb.107.2.665
|
| [51] |
Joshi HC, Chu D, Buxbaum RE, et al. (1985) Tension and compression in the cytoskeleton of PC-12 neurites. J Cell Biol 101: 697–705. doi: 10.1083/jcb.101.3.697
|
| [52] |
Marx KA, Zhou T, Montrone A, et al. (2007) A comparative study of the cytoskeleton binding drugs nocodazole and taxol with a mammalian cell quartz crystal microbalance biosensor: Different dynamic responses and energy dissipation effects. Anal Biochem 361: 77–92. doi: 10.1016/j.ab.2006.09.023
|
| [53] | Arborgh B, Bell P, Brunk U, et al. (1976) The osmotic effect of glutaraldehyde during fixation. A transmission electron microscopy, scanning electron microscopy and cytochemical study. J Ultrastruct Res 56: 339–350. |
| [54] |
Braet F, Rotsch C, Wisse E, et al. (1998) Comparison of fixed and living liver endothelial cells by atomic force microscopy. Appl Phys A-Mater 66: S575. doi: 10.1007/s003390051204
|
| [55] |
Butt HJ, Wolff EK, Gould SAC, et al. (1990) Imaging cells with the atomic force microscope. J Struct Biol 105: 54–61. doi: 10.1016/1047-8477(90)90098-W
|
| [56] |
Dulińska I, Targosz M, Strojny W, et al. (2006) Stiffness of normal and pathological erythrocytes studied by means of atomic force microscopy. J Biochem Bioph Meth 66: 1–11. doi: 10.1016/j.jbbm.2005.11.003
|
| [57] |
Kuznetsova TG, Starodubtseva MN, Yegorenkov NI, et al. (2007) Atomic force microscopy probing of cell elasticity. Micron 38: 824–833. doi: 10.1016/j.micron.2007.06.011
|
| [58] |
Le Grimellec C, Lesniewska E, Cachia C, et al. (1994) Imaging of the membrane surface of MDCK cells by atomic force microscopy. Biophys J 67: 36–41. doi: 10.1016/S0006-3495(94)80490-4
|
| [59] |
Lulevich V, Zink T, Chen H-Y, et al. (2006) Cell mechanics using atomic force microscopy-based single-cell compression. Langmuir 22: 8151–8155. doi: 10.1021/la060561p
|
| [60] |
Mathur AB, Truskey GA, Monty Reichert W (2000) Atomic force and total internal reflection fluorescence microscopy for the study of force transmission in endothelial cells. Biophys J 78: 1725–1735. doi: 10.1016/S0006-3495(00)76724-5
|
| [61] |
Zachee P, Snauwaert J, Vandenberghe P, et al. (1996) Imaging red blood cells with the atomic force microscope. Brit J Haematol 95: 472–481. doi: 10.1111/j.1365-2141.1996.tb08991.x
|
| [62] |
Tsai MA, Waugh RE, Keng PC (1998) Passive mechanical behavior of human Neutrophils: effects of colchicine and paclitaxel. Biophys J 74: 3282–3291. doi: 10.1016/S0006-3495(98)78035-X
|
| [63] |
Rotsch C, Radmacher M (2000) Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: An atomic force microscopy study. Biophys J 78: 520–535. doi: 10.1016/S0006-3495(00)76614-8
|
| [64] | Qiu H, Zhu Y, Sun Z, et al. (2010) Short Communication: Vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107: 615–619. |
| [65] | Wu HW, Kuhn T, Moy VT (1998) Mechanical properties of L929 cells measured by atomic force microscopy: Effects of anticytoskeletal drugs and membrane crosslinking. Scanning 20: 389–397. |
| [66] | Hyo Il Jung Incheol S, Young Mok P, Ke Won K, et al. (1997) Colchicine activates actin polymerization by microtubule depolymerization. Mol Cells 7: 431–437. |
| [67] |
Garland DL (1978) Kinetics and mechanism of colchicine binding to tubulin: evidence for ligand-induced conformational changes. Biochemistry 17: 4266–4272. doi: 10.1021/bi00613a024
|
| [68] |
Spedden E, Staii C (2013) Neuron biomechanics probed by atomic force microscopy. Int J Mol Sci 14: 16124–16140. doi: 10.3390/ijms140816124
|
| [69] |
Spedden E, White JD, Naumova EN, et al. (2012) Elasticity maps of living neurons measured by combined fluorescence and atomic force microscopy. Biophys J 103: 868–877. doi: 10.1016/j.bpj.2012.08.005
|
| [70] |
Canale C, Seghezza S, Vilasi S, et al. (2013) Different effects of Alzheimer's peptide Aβ(1–40) oligomers and fibrils on supported lipid membranes. Biophys Chem 182: 23–29. doi: 10.1016/j.bpc.2013.07.010
|
| [71] |
McLaurin J, Chakrabartty A (1996) Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides: implications for neurotoxicity. J Biol Chem 271: 26482–26489. doi: 10.1074/jbc.271.43.26482
|
| [72] |
Deshpande A, Mina E, Glabe C, et al. (2006) Different conformations of amyloid-β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26: 6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006
|
| [73] |
Blackley HKL, Sanders GHW, Davies MC, et al. (2000) In-situ atomic force microscopy study of β-amyloid fibrillization. J Mol Biol 298: 833–840. doi: 10.1006/jmbi.2000.3711
|
| [74] |
Cizas P, Budvytyte R, Morkuniene R, et al. (2010) Size-dependent neurotoxicity of β-amyloid oligomers. Arch Biochem Biophys 496: 84–92. doi: 10.1016/j.abb.2010.02.001
|
| [75] |
Mastrangelo IA, Ahmed M, Sato T, et al. (2006) High-resolution atomic force microscopy of soluble Aβ42 oligomers. J Mol Biol 358: 106–119. doi: 10.1016/j.jmb.2006.01.042
|
| [76] | Reinert KC, Dunbar RL, Gao W, et al. (2004) Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol 92:199–211. |
| [77] |
Yates EA, Legleiter J (2014) Preparation Protocols of A beta(1-40) Promote the Formation of Polymorphic Aggregates and Altered Interactions with Lipid Bilayers. Biochemistry 53: 7038–7050. doi: 10.1021/bi500792f
|
| [78] |
Demuro A, Mina E, Kayed R, et al. (2005) Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J Biol Chem 280: 17294–17300. doi: 10.1074/jbc.M500997200
|
| [79] |
Demuro A, Parker I, Stutzmann GE (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285: 12463–12468. doi: 10.1074/jbc.R109.080895
|
| [80] |
Kakio A, Nishimoto S-i, Yanagisawa K, et al. (2002) Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41: 7385–7390. doi: 10.1021/bi0255874
|
| [81] |
Kayed R, Sokolov Y, Edmonds B, et al. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279: 46363–46366. doi: 10.1074/jbc.C400260200
|
| [82] |
Kremer JJ, Pallitto MM, Sklansky DJ, et al. (2000) Correlation of β-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry 39: 10309–10318. doi: 10.1021/bi0001980
|
| [83] |
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34: 385–397. doi: 10.1016/S0143-4160(03)00128-3
|
| [84] |
Quist A, Doudevski I, Lin H, et al. (2005) Amyloid ion channels: A common structural link for protein-misfolding disease. P Natl Acad Sci USA 102: 10427–10432. doi: 10.1073/pnas.0502066102
|
| [85] |
Lulevich V, Zimmer CC, Hong H-s, et al. (2010) Single-cell mechanics provides a sensitive and quantitative means for probing amyloid-β peptide and neuronal cell interactions. P Natl Acad Sci USA 107: 13872–13877. doi: 10.1073/pnas.1008341107
|
| [86] | Choi YJ, Chae S, Kim JH, et al. (2013) Neurotoxic amyloid-β oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci Rep 3:1921. |
| [87] |
Lin H, Bhatia R, Lal R (2001) Amyloid-β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15: 2433–2444. doi: 10.1096/fj.01-0377com
|
| [88] | Michaelis ML, Ansar S, Chen Y, et al. (2005) β-amyloid-induced neurodegeneration and protection by structurally diverse microtubule-stabilizing agents. J Pharmocol Exp Ther 312: 659–668. |
| [89] |
Piccini A, Russo C, Gliozzi A, et al. (2005) β-amyloid Is different in normal aging and in Alzheimer disease. J Biol Chem 280: 34186–34192. doi: 10.1074/jbc.M501694200
|
| [90] |
Walsh DM, Hartley DM, Kusumoto Y, et al. (1999) Amyloid β-protein fibrillogenesis - Structure and biological activity of protofibrillar intermediates. J Biol Chem 274: 25945–25952. doi: 10.1074/jbc.274.36.25945
|
| [91] |
Anesti V, Scorrano L (2006) The relationship between mitochondrial shape and function and the cytoskeleton. BBA-Bioenergetics 1757: 692–699. doi: 10.1016/j.bbabio.2006.04.013
|
| [92] | Ball EH, Singer SJ (1982) Mitochondria are associated with microtubules and not with intermediate filaments in cultured fibroblasts. P Natl Acad Sci USA 79: 123–126. |
| [93] | Fuchs F, Prokisch H, Neupert W, et al. (2002) Interaction of mitochondria with microtubules in the filamentous fungus Neurospora crassa. J Cell Sci 115: 1931–1937. |
| [94] | Heggeness MH, Simon M, Singer SJ (1978) Association of mitochondria with microtubules in cultured cells. P Natl Acad Sci USA 75: 3863–3866. |
| [95] |
Shahpasand K, Uemura I, Saito T, et al. (2012) Regulation of mitochondrial transport and inter-microtubule spacing by Tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci 32: 2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012
|
| [96] | De Brabander MJ, Van de Velre RML, Aerts FEM, et al. (1976) The effects of methyl [5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl]carbamate, (R 17934; NSC 238159), a new synthetic antitumoral drug interfering with microtubules, on mammalian cells cultured in vitro. Cancer Res 36: 905–916. |
| [97] |
Bryan J (1972) Vistabline and microtubules: Definition of 3-classes of binding-sites in isolated microtubule crystals. Biochemistry 11: 2611–2616. doi: 10.1021/bi00764a010
|
| [98] |
Hoebeke J, Vannijen G, Debrabander M (1976) Interaction of oncodazole (R17934), new anti-tumoral drug, with rat brain tubulin. Biochem Biophys Res Co 69: 319–324. doi: 10.1016/0006-291X(76)90524-6
|
| [99] |
Owellen RJ, Owens AH, Donigian DW (1972) Binding of vincristine, vistabline and colchicine to tubulin. Biochem Biophys Res Co 47: 685. doi: 10.1016/0006-291X(72)90546-3
|
| [100] |
Ringel I, Sternlicht H (1984) C-13 NMR study of microtubule protein- evidence for a 2nd colchicine site involved in the inhibition of microtubule assembly. Biochemistry 23: 5644–5653. doi: 10.1021/bi00318a040
|
| [101] |
Williams RF, Aivaliotis MJ, Barnes LD, et al. (1983) High performance liquid-chromatographic application of the Hummel and Dreyer method for the determination of colchicine-tubulin binding parameters. J Chromatogr 266: 141–150. doi: 10.1016/S0021-9673(01)90886-6
|
| [102] | Xu K, Schwarz PM, Luduena RF (2002) Interaction of nocodazole with tubulin isotypes. Drug Develop Res 55: 91–96. |
| [103] | Head J, Lee LLY, Field DJ, et al. (1985) Equilibrium and rapid kinetic studies on nocodazole-tubulin interaction. J Biol Chem 260: 1060–1066. |
| [104] | Samson F, Donoso JA, Hellerbettinger I, et al. (1979) Nocodazole action on tubulin assembly, axonal ultrastructure and fast axoplasmic transport. J Pharmacol Exp Ther 208: 411–417. |
| [105] |
Zieve GW, Turnbull D, Mullins JM, et al. (1980) Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole- nocodazole accumulated mitotic cells. Exp Cell Res 126: 397–405. doi: 10.1016/0014-4827(80)90279-7
|
| [106] | Michaelis ML, Ranciat N, Chen Y, et al. (1998) Protection against β-amyloid toxicity in primary neurons by paclitaxel (Taxol). J Neurochem 70: 1623–1627. |
| [107] |
Michaelis ML, Chen YX, Hill S, et al. (2002) Amyloid peptide toxicity and microtubule-stabilizing drugs. J Mol Neurosci 19: 101–105. doi: 10.1007/s12031-002-0018-2
|
| [108] |
Nilsson MR (2004) Techniques to study amyloid fibril formation in vitro. Methods 34: 151–160. doi: 10.1016/j.ymeth.2004.03.012
|
| [109] |
Dahlgren KN, Manelli AM, Stine WB, et al. (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem 277: 32046–32053. doi: 10.1074/jbc.M201750200
|
| [110] |
Burke KA, Yates EA, Legleiter J (2013) Amyloid-forming proteins alter the local mechanical properties of lipid membranes. Biochemistry 52: 808–817. doi: 10.1021/bi301070v
|
| [111] |
Song C, Perides G, Wang D, et al. (2002) β-Amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J Neurochem 83: 828–836. doi: 10.1046/j.1471-4159.2002.01182.x
|
| [112] |
Ma Q-L, Yang F, Rosario ER, et al. (2009) β-amyloid oligomers induce phosphorylation of Tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 29: 9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009
|
| [113] |
Esposito G, De Filippis D, Carnuccio R, et al. (2006) The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. J Mol Med 84: 253–258. doi: 10.1007/s00109-005-0025-1
|
| [114] |
Geula C, Wu CK, Saroff D, et al. (1998) Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med 4: 827–831. doi: 10.1038/nm0798-827
|
| [115] |
Götz J, Chen F, van Dorpe J, et al. (2001) Formation of neurofibrillary tangles in P301L Tau transgenic mice induced by Aβ42 fibrils. Science 293: 1491–1495. doi: 10.1126/science.1062097
|
| [116] |
Ballatore C, Brunden KR, Huryn DM, et al. (2012) Microtubule Stabilizing Agents as Potential Treatment for Alzheimer's Disease and Related Neurodegenerative Tauopathies. J Med Chem 55: 8979–8996. doi: 10.1021/jm301079z
|
| [117] | Lee VMY, Daughenbaugh R, Trojanowski JQ (1994) Microtubule stabilizing drugs for the treatment of Alzheimer's disease. Neurobiol Aging 15: S87–S89. |
| [118] |
Zhang B, Carroll J, Trojanowski JQ, et al. (2012) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged Tau transgenic mice. J Neurosci 32: 3601–3611. doi: 10.1523/JNEUROSCI.4922-11.2012
|
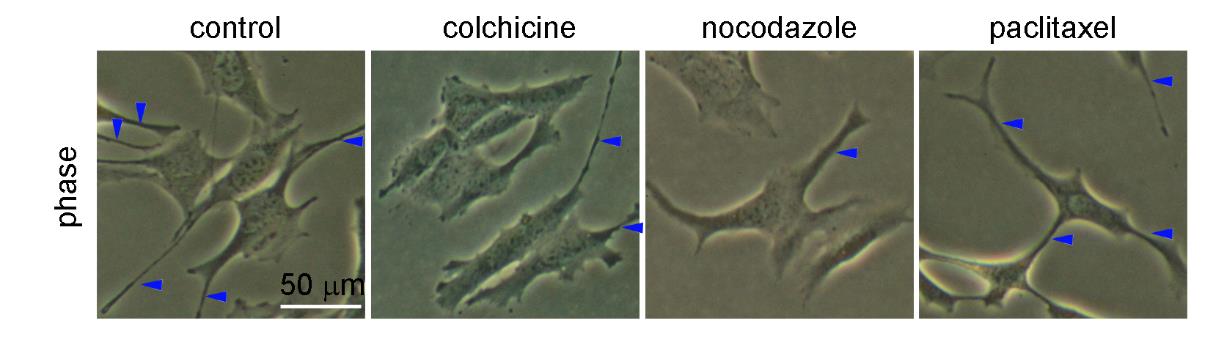
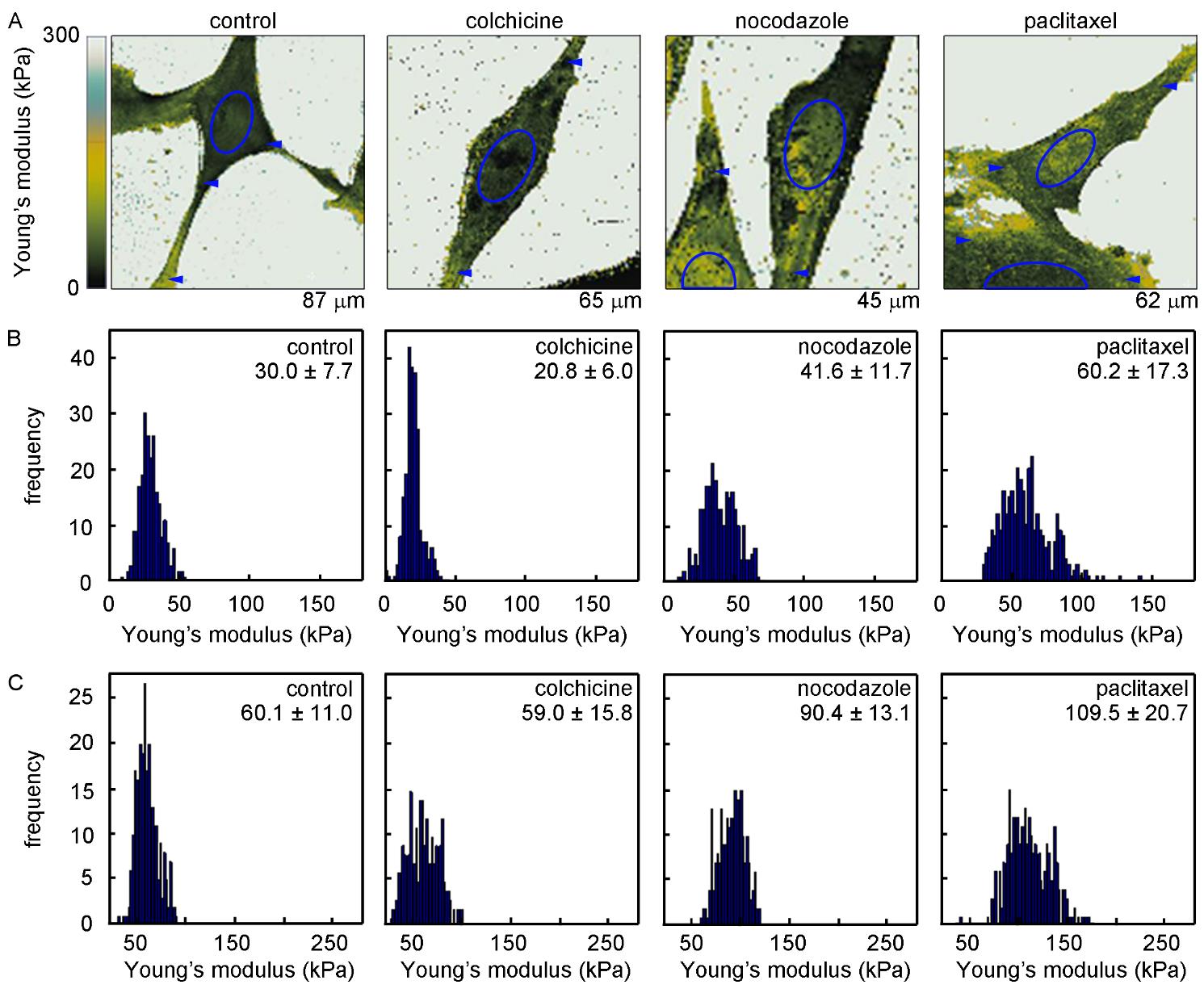
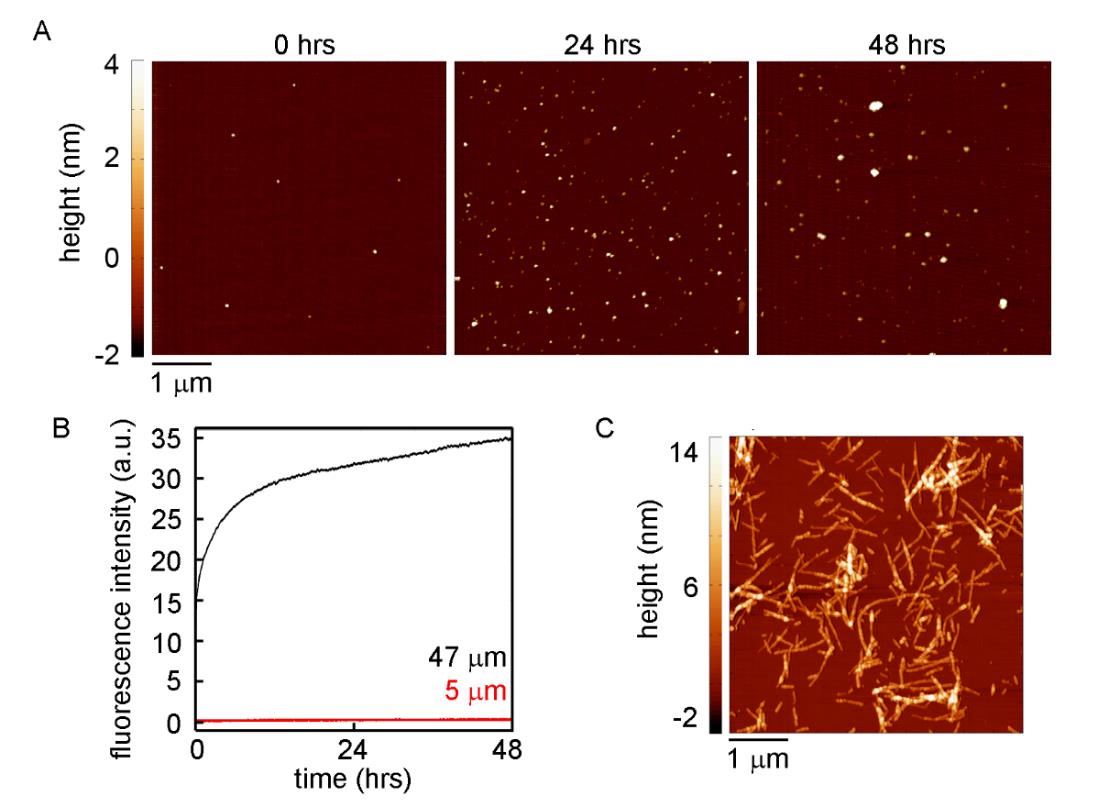
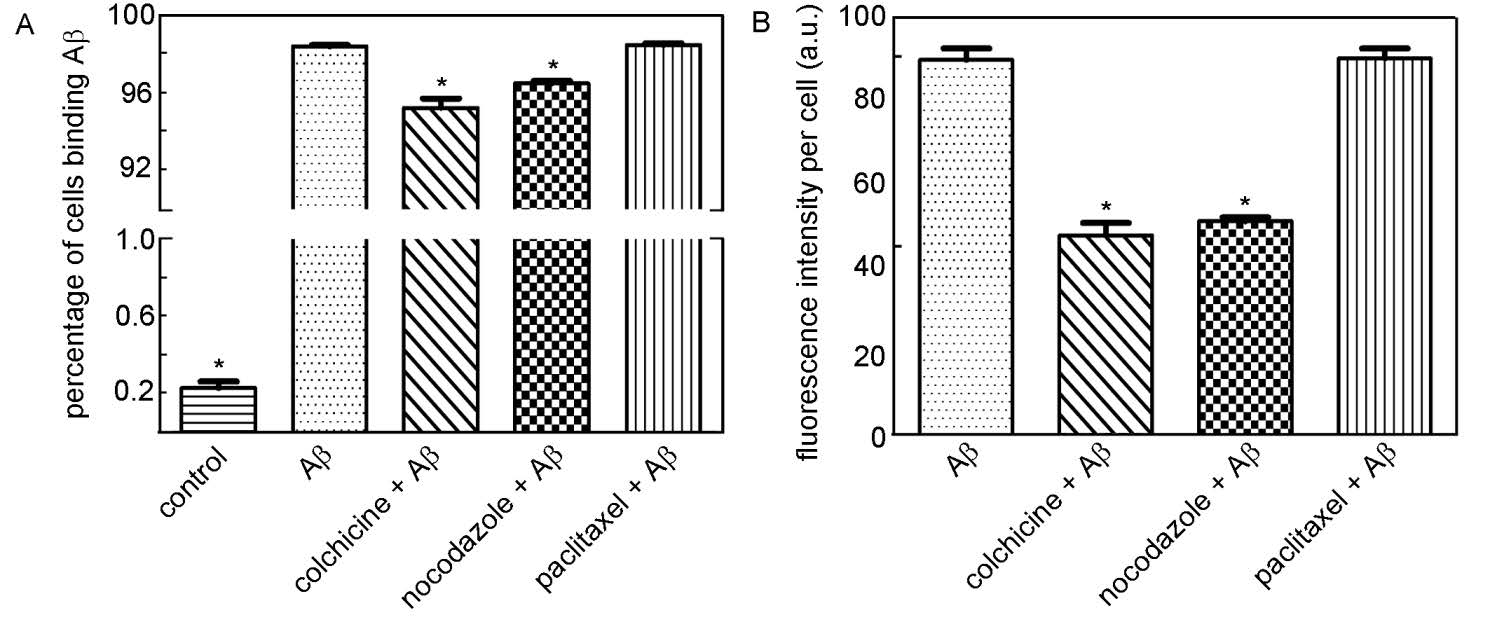
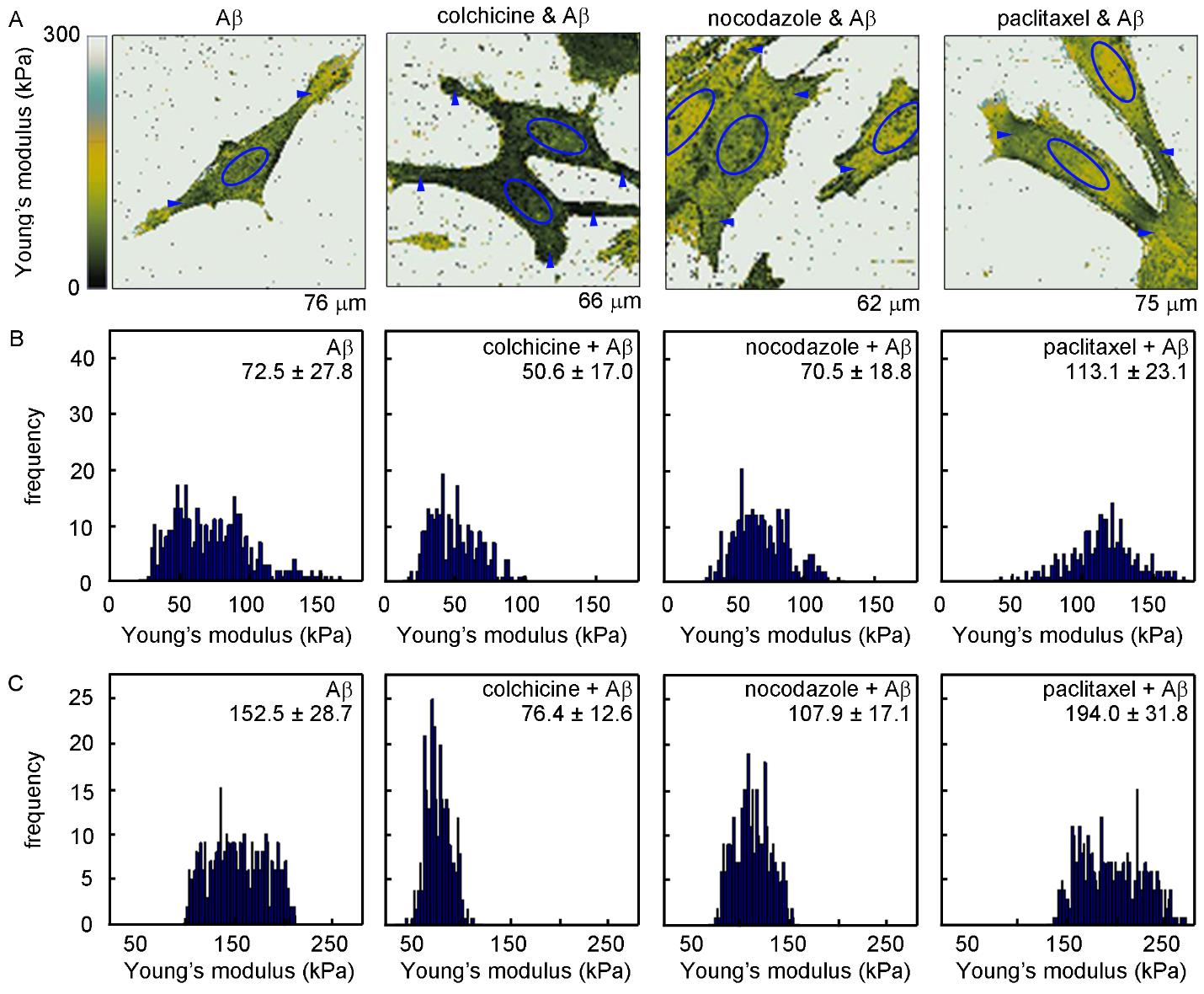
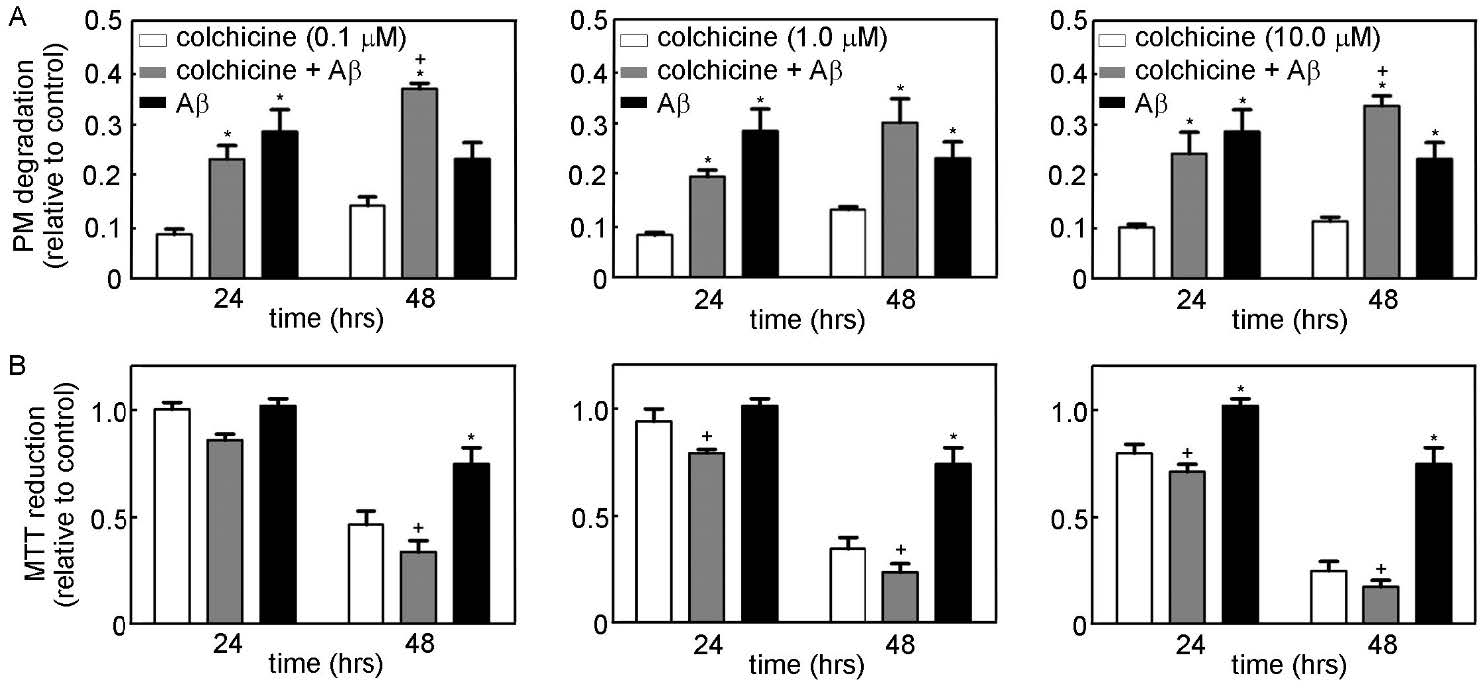
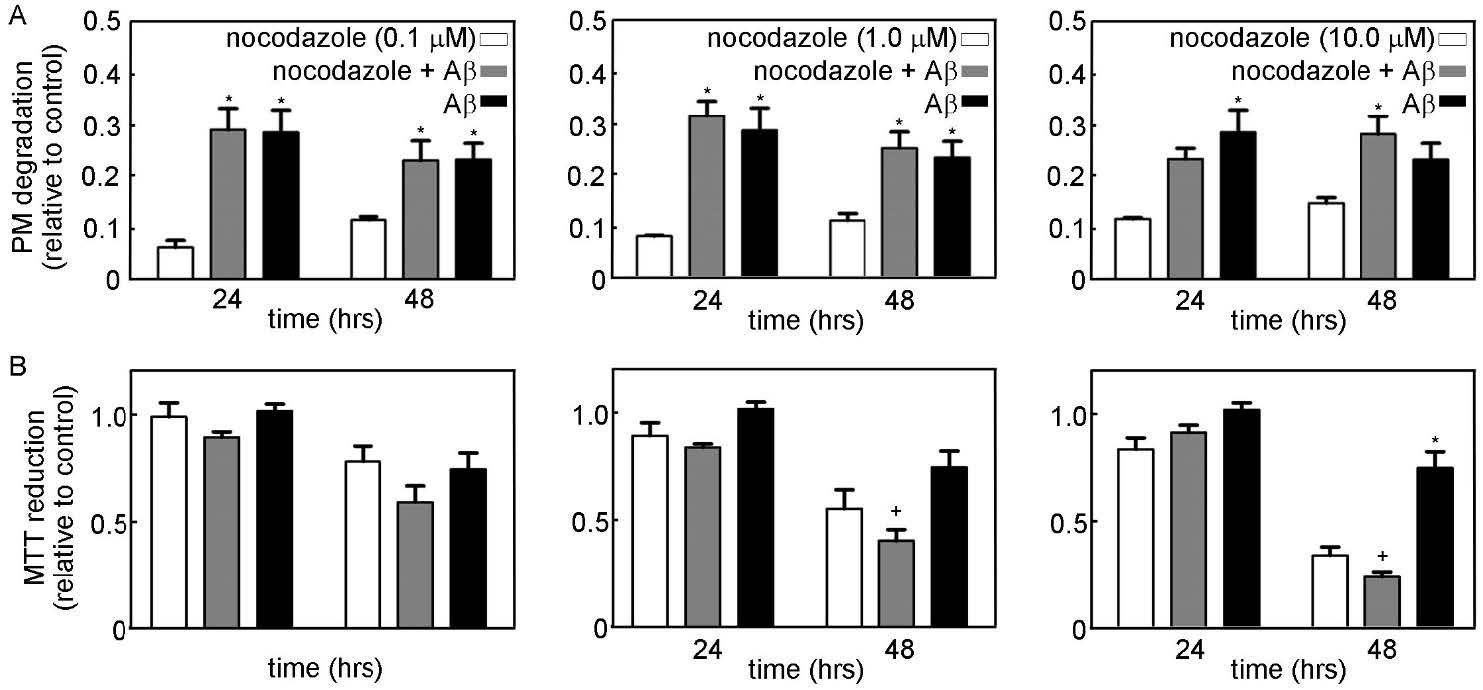
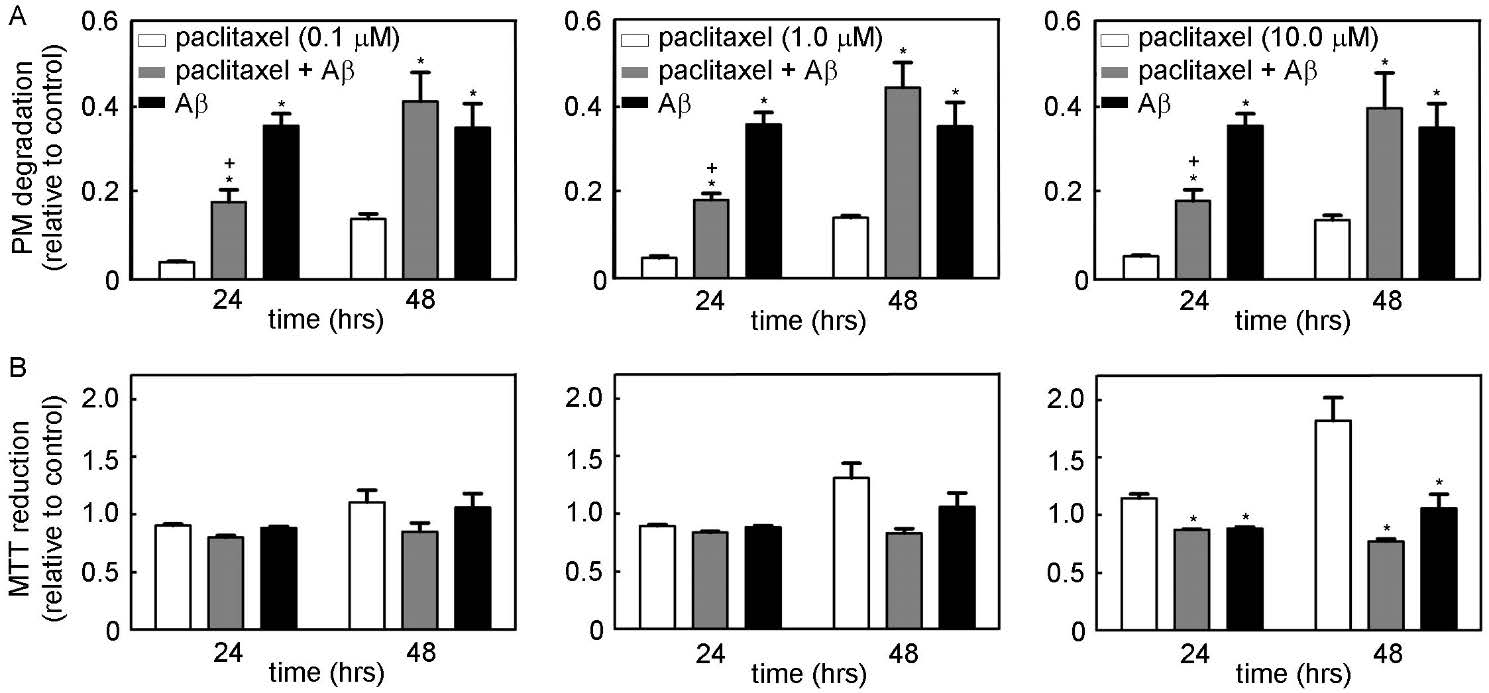
Figures(9) / Tables(1)

Nicole Shamitko-Klingensmith, Jonathan W. Boyd, Justin Legleiter. Microtubule modification influences cellular response to amyloid-β exposure[J]. AIMS Biophysics, 2016, 3(2): 261-285. doi: 10.3934/biophy.2016.2.261







 DownLoad:
DownLoad: