Citation: J.R. Calvo, M.D. Maldonado. The role of melatonin in autoimmune and atopic diseases[J]. AIMS Molecular Science, 2016, 3(2): 158-186. doi: 10.3934/molsci.2016.2.158

| [1] | Lerner AB, Case LD, Lee T, et al. (1958) Isolation of melatonin, the pineal factor that lightens melanocytes. J Am Chem Soc 80: 2587. |
| [2] |
Reiter RJ (1991) Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev 12: 151–180. doi: 10.1210/edrv-12-2-151

|
| [3] |
Barrett P, Bolborea M (2012) Molecular pathways involved in seasonal body weight and reproductive responses governed by melatonin. J Pineal Res 52: 376–388. doi: 10.1111/j.1600-079X.2011.00963.x
|
| [4] |
Stehle JH, Saade A, Rawashdeh O, et al. (2011) A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res 51: 17–43. doi: 10.1111/j.1600-079X.2011.00856.x
|
| [5] |
Hardeland R, Poeggeler B (2003) Non-vertebrate melatonin. J Pineal Res 34: 233–241. doi: 10.1034/j.1600-079X.2003.00040.x
|
| [6] |
Tan DX, Hardeland R, Manchester LC, et al. (2012) Functional roles of melatonin in plants, and perspectives in nutritional and agricultural science. J Exp Bot 63: 577–597. doi: 10.1093/jxb/err256
|
| [7] | Bubenik GA (2008) Thirty four years since the discovery of gastrointestinal melatonin. J Physiol Pharmacol 59 Suppl 2: 33–51. |
| [8] |
Chen CQ, Fichna J, Bashashati M, et al. (2011) Distribution, function and physiological role of melatonin in the lower gut. World J Gastroenterol 17: 3888–3898. doi: 10.3748/wjg.v17.i34.3888
|
| [9] |
Hardeland R, Cardinali DP, Srinivasan V, et al. (2011) Melatonin-a pleiotropic, orchestrating regulator molecule. Prog Neurobiol 93: 350–384. doi: 10.1016/j.pneurobio.2010.12.004
|
| [10] |
Shi L, Li N, Bo L, et al. (2013) Melatonin and hypothalamic-pituitary-gonadal axis. Curr Med Chem 20: 2017–2031. doi: 10.2174/09298673113209990114
|
| [11] |
Carrillo-Vico A, Lardone PJ, Alvarez-Sanchez N, et al. (2013) Melatonin: buffering the immune system. Int J Mol Sci 14: 8638–8683. doi: 10.3390/ijms14048638
|
| [12] |
Calvo JR, Gonzalez-Yanes C, Maldonado MD (2013) The role of melatonin in the cells of the innate immunity: a review. J Pineal Res 55: 103–120. doi: 10.1111/jpi.12075
|
| [13] |
Slominski A, Tobin DJ, Zmijewski MA, et al. (2008) Melatonin in the skin: synthesis, metabolism and functions. Trends Endocrinol Metab 19: 17–24. doi: 10.1016/j.tem.2007.10.007
|
| [14] | Poeggeler B, Saarela S, Reiter RJ, et al. (1994) Melatonin-a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro. Ann N Y Acad Sci 738: 419–420. |
| [15] |
Ressmeyer AR, Mayo JC, Zelosko V, et al. (2003) Antioxidant properties of the melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK): scavenging of free radicals and prevention of protein destruction. Redox Rep 8: 205–213. doi: 10.1179/135100003225002709
|
| [16] |
Hardeland R, Tan DX, Reiter RJ (2009) Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J Pineal Res 47: 109–126. doi: 10.1111/j.1600-079X.2009.00701.x
|
| [17] |
Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 51: 1–16. doi: 10.1111/j.1600-079X.2011.00916.x
|
| [18] |
Galano A, Tan DX, Reiter RJ (2013) On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J Pineal Res 54: 245–257. doi: 10.1111/jpi.12010
|
| [19] |
Lissoni P, Barni S, Tancini G, et al. (1993) A study of the mechanisms involved in the immunostimulatory action of the pineal hormone in cancer patients. Oncology 50: 399–402. doi: 10.1159/000227218
|
| [20] |
Sanchez-Hidalgo M, Lee M, de la Lastra CA, et al. (2012) Melatonin inhibits cell proliferation and induces caspase activation and apoptosis in human malignant lymphoid cell lines. J Pineal Res 53: 366–373. doi: 10.1111/j.1600-079X.2012.01006.x
|
| [21] |
Reiter RJ (1992) The ageing pineal gland and its physiological consequences. Bioessays 14: 169–175. doi: 10.1002/bies.950140307
|
| [22] |
Poeggeler B (2005) Melatonin, aging, and age-related diseases: perspectives for prevention, intervention, and therapy. Endocrine 27: 201–212. doi: 10.1385/ENDO:27:2:201
|
| [23] |
Guerrero JM, Reiter RJ (1992) A brief survey of pineal gland-immune system interrelationships. Endocr Res 18: 91–113. doi: 10.1080/07435809209035401
|
| [24] |
Dubocovich ML (1995) Melatonin receptors: are there multiple subtypes? Trends Pharmacol Sci 16: 50–56. doi: 10.1016/S0165-6147(00)88978-6
|
| [25] |
Maldonado MD, Garcia-Moreno H, Calvo JR (2013) Melatonin protects mast cells against cytotoxicity mediated by chemical stimuli PMACI: possible clinical use. J Neuroimmunol 262: 62–65. doi: 10.1016/j.jneuroim.2013.06.013
|
| [26] |
Benitez-King G, Huerto-Delgadillo L, Anton-Tay F (1993) Binding of 3H-melatonin to calmodulin. Life Sci 53: 201–207. doi: 10.1016/0024-3205(93)90670-X
|
| [27] |
Gitto E, Pellegrino S, Gitto P, et al. (2009) Oxidative stress of the newborn in the pre- and postnatal period and the clinical utility of melatonin. J Pineal Res 46: 128–139. doi: 10.1111/j.1600-079X.2008.00649.x
|
| [28] | Korkmaz A, Reiter RJ, Topal T, et al. (2009) Melatonin: an established antioxidant worthy of use in clinical trials. Mol Med 15: 43–50. |
| [29] |
Blalock JE (2005) The immune system as the sixth sense. J Intern Med 257: 126–138. doi: 10.1111/j.1365-2796.2004.01441.x
|
| [30] |
Skwarlo-Sonta K, Majewski P, Markowska M, et al. (2003) Bidirectional communication between the pineal gland and the immune system. Can J Physiol Pharmacol 81: 342–349. doi: 10.1139/y03-026
|
| [31] |
Markus RP, Ferreira ZS, Fernandes PA, et al. (2007) The immune-pineal axis: a shuttle between endocrine and paracrine melatonin sources. Neuroimmunomodulation 14: 126–133. doi: 10.1159/000110635
|
| [32] |
Nelson RJ, Demas GE, Klein SL, et al. (1995) The influence of season, photoperiod, and pineal melatonin on immune function. J Pineal Res 19: 149–165. doi: 10.1111/j.1600-079X.1995.tb00184.x
|
| [33] | Nelson RJ, Drazen DL (2000) Melatonin mediates seasonal changes in immune function. Ann N Y Acad Sci 917: 404–415. |
| [34] |
Pontes GN, Cardoso EC, Carneiro-Sampaio MM, et al. (2006) Injury switches melatonin production source from endocrine (pineal) to paracrine (phagocytes) - melatonin in human colostrum and colostrum phagocytes. J Pineal Res 41: 136–141. doi: 10.1111/j.1600-079X.2006.00345.x
|
| [35] |
Markus RP, Cecon E, Pires-Lapa MA (2013) Immune-pineal axis: nuclear factor kappaB (NF-kB) mediates the shift in the melatonin source from pinealocytes to immune competent cells. Int J Mol Sci 14: 10979–10997. doi: 10.3390/ijms140610979
|
| [36] |
De Oliveira TD, Levandovski RM, Custodio de Souza IC, et al. (2013) The concept of the immune-pineal axis tested in patients undergoing an abdominal hysterectomy. Neuroimmunomodulation 20: 205–212. doi: 10.1159/000347160
|
| [37] | Slominski A, Wortsman J (2000) Neuroendocrinology of the skin. Endocr Rev 21: 457–487. |
| [38] |
Slominski AT, Zmijewski MA, Skobowiat C, et al. (2012) Sensing the environment: regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv Anat Embryol Cell Biol 212: 1–115. doi: 10.1007/978-3-642-19683-6_1
|
| [39] | Slominski A, Wortsman J, Luger T, et al. (2000) Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev 80: 979–1020. |
| [40] | Slominski A, Pisarchik A, Semak I, et al. (2002) Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J 16: 896–898. |
| [41] |
Slominski A, Baker J, Rosano TG, et al. (1996) Metabolism of serotonin to N-acetylserotonin, melatonin, and 5-methoxytryptamine in hamster skin culture. J Biol Chem 271: 12281–12286. doi: 10.1074/jbc.271.21.12281
|
| [42] |
Slominski A, Wortsman J, Tobin DJ (2005) The cutaneous serotoninergic/melatoninergic system: securing a place under the sun. FASEB J 19: 176–194. doi: 10.1096/fj.04-2079rev
|
| [43] |
Slominski A, Pisarchik A, Zbytek B, et al. (2003) Functional activity of serotoninergic and melatoninergic systems expressed in the skin. J Cell Physiol 196: 144–153. doi: 10.1002/jcp.10287
|
| [44] |
Slominski A, Fischer TW, Zmijewski MA, et al. (2005) On the role of melatonin in skin physiology and pathology. Endocrine 27: 137–148. doi: 10.1385/ENDO:27:2:137
|
| [45] |
Fischer TW, Sweatman TW, Semak I, et al. (2006) Constitutive and UV-induced metabolism of melatonin in keratinocytes and cell-free systems. FASEB J 20: 1564–1566. doi: 10.1096/fj.05-5227fje
|
| [46] |
Kim T-K, Kleszczynski K, Janjetovic Z, et al. (2013) Metabolism of melatonin and biological activity of intermediates of melatoninergic pathway in human skin cells. FASEB J 27: 2742–2755. doi: 10.1096/fj.12-224691
|
| [47] |
Kim T-K, Lin Z, Tidwell WJ, et al. (2015) Melatonin and its metabolites accumulate in the human epidermis in vivo and inhibit proliferation and tyrosinase activity in epidermal melanocytes in vitro. Mol Cell Edocrinology 404: 1–8. doi: 10.1016/j.mce.2014.07.024
|
| [48] |
Kim T-K, Lin Z, Li W, et al. (2015) N1-Acetyl-5-Methoxykynuramine (AMK) is produced in the human epidermis and shows antiproliferative effects. Endocrinology 156: 1630–1636. doi: 10.1210/en.2014-1980
|
| [49] |
Slominski AT, Kleszczyński K, Semak I, et al. (2014) Local melatoninergic system as the protector of skin integrity. Int J Mol Sci 15: 17705–17732. doi: 10.3390/ijms151017705
|
| [50] |
Calvo JR, Rafii-El-Idrissi M, Pozo D, et al. (1995) Immunomodulatory role of melatonin: specific binding sites in human and rodent lymphoid cells. J Pineal Res 18: 119–126. doi: 10.1111/j.1600-079X.1995.tb00149.x
|
| [51] | Maldonado MD, Mora-Santos M, Naji L, et al. (2010) Evidence of melatonin synthesis and release by mast cells. Possible modulatory role on inflammation. Pharmacol Res 62: 282–287. |
| [52] |
Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, et al. (2012) Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol 351: 152–166. doi: 10.1016/j.mce.2012.01.004
|
| [53] |
Yu ZH, Yuan H, Lu Y, et al. (1991) [125I]iodomelatonin binding sites in spleens of birds and mammals. Neurosci Lett 125: 175–178. doi: 10.1016/0304-3940(91)90021-K
|
| [54] |
Pang CS, Pang SF (1992) High affinity specific binding of 2-[125I]iodomelatonin by spleen membrane preparations of chicken. J Pineal Res 12: 167–173. doi: 10.1111/j.1600-079X.1992.tb00044.x
|
| [55] |
Poon AM, Wang XL, Pang SF (1993) Characteristics of 2-[125I]iodomelatonin binding sites in the pigeon spleen and modulation of binding by guanine nucleotides. J Pineal Res 14: 169–177. doi: 10.1111/j.1600-079X.1993.tb00499.x
|
| [56] |
Lopez-Gonzalez MA, Martin-Cacao A, Calvo JR, et al. (1993) Specific binding of 2-[125I]melatonin by partially purified membranes of rat thymus. J Neuroimmunol 45: 121–126. doi: 10.1016/0165-5728(93)90171-T
|
| [57] |
Rafii-El-Idrissi M, Calvo JR, Pozo D, et al. (1995) Specific binding of 2-[125I]iodomelatonin by rat splenocytes: characterization and its role on regulation of cyclic AMP production. J Neuroimmunol 57: 171–178. doi: 10.1016/0165-5728(94)00182-N
|
| [58] | Garcia-Perganeda A, Pozo D, Guerrero JM, et al. (1997) Signal transduction for melatonin in human lymphocytes: involvement of a pertussis toxin-sensitive G protein. J Immunol 159: 3774–3781. |
| [59] |
Garcia-Perganeda A, Guerrero JM, Rafii-El-Idrissi M, et al. (1999) Characterization of membrane melatonin receptor in mouse peritoneal macrophages: inhibition of adenylyl cyclase by a pertussis toxin-sensitive G protein. J Neuroimmunol 95: 85–94. doi: 10.1016/S0165-5728(98)00268-9
|
| [60] |
Lopez-Gonzalez MA, Calvo JR, Osuna C, et al. (1992) Interaction of melatonin with human lymphocytes: evidence for binding sites coupled to potentiation of cyclic AMP stimulated by vasoactive intestinal peptide and activation of cyclic GMP. J Pineal Res 12: 97–104. doi: 10.1111/j.1600-079X.1992.tb00034.x
|
| [61] |
Poon AM, Pang SF (1992) 2[125I]iodomelatonin binding sites in spleens of guinea pigs. Life Sci 50: 1719–1726. doi: 10.1016/0024-3205(92)90427-Q
|
| [62] | Dubocovich ML, Masana MI, Benloucif S (1999) Molecular pharmacology and function of melatonin receptor subtypes. Adv Exp Med Biol 460: 181–190. |
| [63] | Pozo D, Delgado M, Fernandez-Santos JM, et al. (1997) Expression of the Mel1a-melatonin receptor mRNA in T and B subsets of lymphocytes from rat thymus and spleen. FASEB J 11: 466–473. |
| [64] |
Garcia-Maurino S, Pozo D, Calvo JR, et al. (2000) Correlation between nuclear melatonin receptor expression and enhanced cytokine production in human lymphocytic and monocytic cell lines. J Pineal Res 29: 129–137. doi: 10.1034/j.1600-079X.2000.290301.x
|
| [65] |
Carrillo-Vico A, Garcia-Perganeda A, Naji L, et al. (2003) Expression of membrane and nuclear melatonin receptor mRNA and protein in the mouse immune system. Cell Mol Life Sci 60: 2272–2278. doi: 10.1007/s00018-003-3207-4
|
| [66] | Carrillo-Vico A, Garcia-Maurino S, Calvo JR, et al. (2003) Melatonin counteracts the inhibitory effect of PGE2 on IL-2 production in human lymphocytes via its mt1 membrane receptor. FASEB J 17: 755–757. |
| [67] |
Pozo D, Garcia-Maurino S, Guerrero JM, et al. (2004) mRNA expression of nuclear receptor RZR/RORalpha, melatonin membrane receptor MT, and hydroxindole-O-methyltransferase in different populations of human immune cells. J Pineal Res 37: 48–54. doi: 10.1111/j.1600-079X.2004.00135.x
|
| [68] | Kallen JA, Schlaeppi J-M, Bitsch F, et al. (2002) X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha. Structure 10: 1697–707. |
| [69] |
Kallen J, Schlaeppi J-M, Bitsch F, et al. (2004) Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem 279: 14033–14038. doi: 10.1074/jbc.M400302200
|
| [70] |
Slominski AT, Kim T-K, Takeda Y, et al. (2014) RORα and ROR γ are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20-hydroxy- and 20,23-dihydroxyvitamin D. FASEB J 28: 2775–2789. doi: 10.1096/fj.13-242040
|
| [71] |
Janeway Jr. CA (1992) The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today 13: 11–16. doi: 10.1016/0167-5699(92)90198-G
|
| [72] |
Kay AB (2001) Allergy and allergic diseases. First of two parts. N Engl J Med 344: 30–37. doi: 10.1056/NEJM200101043440106
|
| [73] | Kay AB (2001) Allergy and allergic diseases. Second of two parts. N Engl J Med 344: 109–113. |
| [74] |
Ono SJ (2000) Molecular genetics of allergic diseases. Annu Rev Immunol 18: 347–366. doi: 10.1146/annurev.immunol.18.1.347
|
| [75] |
Galli SJ, Tsai M, Piliponsky AM (2008) The development of allergic inflammation. Nature 454: 445–454. doi: 10.1038/nature07204
|
| [76] |
Rothenberg ME, Daeron M (2009) Hypersensitivity and allergy: from mice to men. Curr Opin Immunol 21: 658–659. doi: 10.1016/j.coi.2009.10.001
|
| [77] |
Fontenot AP, Peebles Jr. RS (2013) Allergy and hypersensitivity. Curr Opin Immunol 25: 736–737. doi: 10.1016/j.coi.2013.10.007
|
| [78] |
Davidson A, Diamond B (2001) Autoimmune diseases. N Engl J Med 345: 340–350. doi: 10.1056/NEJM200108023450506
|
| [79] |
Parish IA, Heath WR (2008) Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunol Cell Biol 86: 146–152. doi: 10.1038/sj.icb.7100161
|
| [80] |
Goodnow CC, Sprent J, Fazekas de St GB, et al. (2005) Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 435: 590–597. doi: 10.1038/nature03724
|
| [81] |
Pascual V, Chaussabel D, Banchereau J (2010) A genomic approach to human autoimmune diseases. Annu Rev Immunol 28: 535–571. doi: 10.1146/annurev-immunol-030409-101221
|
| [82] |
Goodnow CC (2007) Multistep pathogenesis of autoimmune disease. Cell 130: 25–35. doi: 10.1016/j.cell.2007.06.033
|
| [83] |
Leung DY, Boguniewicz M, Howell MD, et al. (2004) New insights into atopic dermatitis. J Clin Invest 113: 651–657. doi: 10.1172/JCI21060
|
| [84] |
Grewe M, Bruijnzeel-Koomen CA, Schopf E, et al. (1998) A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol Today 19: 359–361. doi: 10.1016/S0167-5699(98)01285-7
|
| [85] |
Miraglia del Giudice M, Decimo F, Leonardi S, et al. (2006) Immune dysregulation in atopic dermatitis. Allergy Asthma Proc 27: 451–455. doi: 10.2500/aap.2006.27.2887
|
| [86] |
Leonardi S, Miraglia del Giudice M, La Rosa M, et al. (2007) Atopic disease, immune system, and the environment. Allergy Asthma Proc 28: 410–417. doi: 10.2500/aap.2007.28.2954
|
| [87] | Schwarz W, Birau N, Hornstein OP, et al. (1988) Alterations of melatonin secretion in atopic eczema. Acta Derm Venereol 68: 224–229. |
| [88] |
Kimata H (2007) Elevation of salivary melatonin levels by viewing a humorous film in patients with atopic eczema. Horm Metab Res 39: 310–311. doi: 10.1055/s-2007-973815
|
| [89] |
Munoz-Hoyos A, Espin-Quirantes C, Molina-Carballo A, et al. (2007) Neuroendocrine and circadian aspects (melatonin and beta-endorphin) of atopic dermatitis in the child. Pediatr Allergy Immunol 18: 679–686. doi: 10.1111/j.1399-3038.2007.00574.x
|
| [90] |
Kim TH, Jung JA, Kim GD, et al. (2009) Melatonin inhibits the development of 2,4-dinitrofluorobenzene-induced atopic dermatitis-like skin lesions in NC/Nga mice. J Pineal Res 47: 324–329. doi: 10.1111/j.1600-079X.2009.00718.x
|
| [91] | Majewska M, Zajac K, Zemelka M, et al. (2007) Influence of melatonin and its precursor L-tryptophan on Th1 dependent contact hypersensitivity. J Physiol Pharmacol 58 Suppl 6: 125–132. |
| [92] | Suke SG, Pathak R, Ahmed RS, et al. (2008) Melatonin treatment prevents modulation of cell-mediated immune response induced by propoxur in rats. Indian J Biochem Biophys 45: 278–281. |
| [93] | Shandra OO (2014) Effect of deltaran and melatonin on immune system in rats with experimental contact dermatitis. Fiziol Zh 60: 78–83. |
| [94] | Kilanczyk E, Bryszewska M (2003) The effect of melatonin on antioxidant enzymes in human diabetic skin fibroblasts. Cell Mol Biol Lett 8: 333–336. |
| [95] |
Fischer TW, Zbytek B, Sayre RM, et al. (2006) Melatonin increases survival of HaCaT keratinocytes by suppressing UV-induced apoptosis. J Pineal Res 40: 18–26. doi: 10.1111/j.1600-079X.2005.00273.x
|
| [96] |
Marseglia L, D’Angelo G, Manti S, et al. (2014) Melatonin and atopy: role in atopic dermatitis and asthma. Int J Mol Sci 15: 13482–13493. doi: 10.3390/ijms150813482
|
| [97] |
Arnetz BB, Berg M (1996) Melatonin and adrenocorticotropic hormone levels in video display unit workers during work and leisure. J Occup Env Med 38: 1108–1110. doi: 10.1097/00043764-199611000-00010
|
| [98] |
Kimata H (2004) Reduction of allergen-specific IgE production by laughter. Eur J Clin Invest 34: 76–77. doi: 10.1111/j.1365-2362.2004.01294.x
|
| [99] | Chang Y-S, Lin M-H, Lee J-H, et al. (2015) Melatonin supplementation for children with atopic dermatitis and sleep disturbance: a randomized clinical trial. JAMA Pediatr 170: 1–8. |
| [100] |
Maddox L, Schwartz DA (2002) The pathophysiology of asthma. Annu Rev Med 53: 477–498. doi: 10.1146/annurev.med.53.082901.103921
|
| [101] |
Sutherland ER (2005) Nocturnal asthma. J Allergy Clin Immunol 116: 1179–1186. doi: 10.1016/j.jaci.2005.09.028
|
| [102] | Karasu-Minareci E, Kaya Y, Belgin YF (2012) The achilles heel in melatonin: asthma. Iran J Allergy Asthma Immunol 11: 246–252. |
| [103] |
Bowler RP, Crapo JD (2002) Oxidative stress in allergic respiratory diseases. J Allergy Clin Immunol 110: 349–356. doi: 10.1067/mai.2002.126780
|
| [104] |
Sackesen C, Ercan H, Dizdar E, et al. (2008) A comprehensive evaluation of the enzymatic and nonenzymatic antioxidant systems in childhood asthma. J Allergy Clin Immunol 122: 78–85. doi: 10.1016/j.jaci.2008.03.035
|
| [105] |
Chipps BE, Zeiger RS, Borish L, et al. (2012) Key findings and clinical implications from The Epidemiology and Natural History of Asthma: Outcomes and Treatment Regimens (TENOR) study. J Allergy Clin Immunol 130: 332–342. doi: 10.1016/j.jaci.2012.04.014
|
| [106] | Fei GH, Liu RY, Zhang ZH, et al. (2003) Relationships between melatonin and cortisol and the status of disease in patients with bronchial asthma. Zhonghua Jie He He Hu Xi Za Zhi 26: 679–682. |
| [107] | Fei GH, Liu RY, Zhang ZH, et al. (2004) Alterations in circadian rhythms of melatonin and cortisol in patients with bronchial asthma. Acta Pharmacol Sin 25: 651–656. |
| [108] | Evsiukova E V (1999) Melatonin effects on functional activity of platelets in patients with bronchial asthma. Ter Arkh 71: 35–37. |
| [109] | Kos-Kudla B, Ostrowska Z, Marek B, et al. (2002) Circadian rhythm of melatonin in postmenopausal asthmatic women with hormone replacement therapy. Neuro Endocrinol Lett 23: 243–248. |
| [110] |
Evsyukova H V (1999) The role of melatonin in pathogenesis of aspirin-sensitive asthma. Eur J Clin Invest 29: 563–567. doi: 10.1046/j.1365-2362.1999.00479.x
|
| [111] |
Evsyukova H V (2011) Expression of melatonin in platelets of patients with aspirin-induced asthma. Eur J Clin Invest 41: 781–784. doi: 10.1111/j.1365-2362.2011.02484.x
|
| [112] | Evsiukova E V, Okuneva EI, Zubzhitskaia LB, et al. (2008) Melatonin expression in nasal polyps in patients with asthmatic triad. Arkh Patol 70: 33–35. |
| [113] | Evsyukova H V (2002) Aspirin-sensitive asthma due to diffuse neuroendocrine system pathology. Neuro Endocrinol Lett 23: 281–285. |
| [114] |
Sutherland ER, Martin RJ, Ellison MC, et al. (2002) Immunomodulatory effects of melatonin in asthma. Am J Respir Crit Care Med 166: 1055–1061. doi: 10.1164/rccm.200204-356OC
|
| [115] |
Sutherland ER, Ellison MC, Kraft M, et al. (2003) Elevated serum melatonin is associated with the nocturnal worsening of asthma. J Allergy Clin Immunol 112: 513–517. doi: 10.1016/S0091-6749(03)01717-2
|
| [116] |
Gumral N, Caliskan S, Ozguner F, et al. (2009) Melatonin levels and enzymatic antioxidant defense system decrease in blood of patients with bronchial asthma. Toxicol Ind Heal 25: 411–416. doi: 10.1177/0748233709106625
|
| [117] |
Gumral N, Naziroglu M, Ongel K, et al. (2009) Antioxidant enzymes and melatonin levels in patients with bronchial asthma and chronic obstructive pulmonary disease during stable and exacerbation periods. Cell Biochem Funct 27: 276–283. doi: 10.1002/cbf.1569
|
| [118] |
Campos FL, da Silva-Junior FP, de V B, et al. (2004) Melatonin improves sleep in asthma: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med 170: 947–951. doi: 10.1164/rccm.200404-488OC
|
| [119] |
Martins E, Ligeiro de Oliveira AP, Fialho de Araujo AM, et al. (2001) Melatonin modulates allergic lung inflammation. J Pineal Res 31: 363–369. doi: 10.1034/j.1600-079X.2001.310412.x
|
| [120] |
Luo F, Liu X, Li S, et al. (2004) Melatonin promoted chemotaxins expression in lung epithelial cell stimulated with TNF-alpha. Respir Res 5: 20–28. doi: 10.1186/1465-9921-5-20
|
| [121] | Wang YT, Chen SL, Xu SY (2004) Effect of melatonin on the expression of nuclear factor-kappa B and airway inflammation in asthmatic rats. Zhonghua Er Ke Za Zhi 42: 94–97. |
| [122] | Zhou L, Qian ZX, Li F, et al. (2007) The effect of melatonin on the regulation of collagen accumulation and matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 mRNA and protein in a murine model of chronic asthma. Zhonghua Jie He He Hu Xi Za Zhi 30: 527–532. |
| [123] |
Shin IS, Park JW, Shin NR, et al. (2014) Melatonin inhibits MUC5AC production via suppression of MAPK signaling in human airway epithelial cells. J Pineal Res 56: 398–407. doi: 10.1111/jpi.12127
|
| [124] |
Rioux JD, Abbas AK (2005) Paths to understanding the genetic basis of autoimmune disease. Nature 435: 584–589. doi: 10.1038/nature03723
|
| [125] |
Choy EH, Panayi GS (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 344: 907–916. doi: 10.1056/NEJM200103223441207
|
| [126] |
Imboden JB (2009) The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 4: 417–434. doi: 10.1146/annurev.pathol.4.110807.092254
|
| [127] |
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365: 2205–2219. doi: 10.1056/NEJMra1004965
|
| [128] |
Kalpakcioglu B, Senel K (2009) The role of melatonin in rheumatic diseases. Infect Disord Drug Targets 9: 453–456. doi: 10.2174/187152609788922546
|
| [129] |
Sanchez-Barcelo EJ, Mediavilla MD, Tan DX, et al. (2010) Clinical uses of melatonin: evaluation of human trials. Curr Med Chem 17: 2070–2095. doi: 10.2174/092986710791233689
|
| [130] |
Hansson I, Holmdahl R, Mattsson R (1990) Constant darkness enhances autoimmunity to type II collagen and exaggerates development of collagen-induced arthritis in DBA/1 mice. J Neuroimmunol 27: 79–84. doi: 10.1016/0165-5728(90)90139-E
|
| [131] | Hansson I, Holmdahl R, Mattsson R (1993) Pinealectomy ameliorates collagen II-induced arthritis in mice. Clin Exp Immunol 92: 432–436. |
| [132] |
Hansson I, Holmdahl R, Mattsson R (1992) The pineal hormone melatonin exaggerates development of collagen-induced arthritis in mice. J Neuroimmunol 39: 23–30. doi: 10.1016/0165-5728(92)90171-G
|
| [133] |
Mattsson R, Hannsson I, Holmdahl R (1994) Pineal gland in autoimmunity: melatonin-dependent exaggeration of collagen-induced arthritis in mice. Autoimmunity 17: 83–86. doi: 10.3109/08916939409014661
|
| [134] |
Jimenez-Caliani AJ, Jimenez-Jorge S, Molinero P, et al. (2005) Dual effect of melatonin as proinflammatory and antioxidant in collagen-induced arthritis in rats. J Pineal Res 38: 93–99. doi: 10.1111/j.1600-079X.2004.00175.x
|
| [135] | Cano P, Cardinali DP, Chacon F, et al. (2002) Nighttime changes in norepinephrine and melatonin content and serotonin turnover in pineal glands of young and old rats injected with Freund’s adjuvant. Neuro Endocrinol Lett 23: 49–53. |
| [136] |
Bang J, Chang HW, Jung HR, et al. (2012) Melatonin attenuates clock gene cryptochrome1, which may aggravate mouse anti-type II collagen antibody-induced arthritis. Rheumatol Int 32: 379–385. doi: 10.1007/s00296-010-1641-9
|
| [137] |
Chen Q, Wei W (2002) Effects and mechanisms of melatonin on inflammatory and immune responses of adjuvant arthritis rat. Int Immunopharmacol 2: 1443–1449. doi: 10.1016/S1567-5769(02)00088-7
|
| [138] |
Cutolo M, Balleari E, Giusti M, et al. (1988) Sex hormone status of male patients with rheumatoid arthritis: evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheum 31: 1314–1317. doi: 10.1002/art.1780311015
|
| [139] |
Valenti S, Giusti M (2002) Melatonin participates in the control of testosterone secretion from rat testis: an overview of our experience. Ann N Y Acad Sci 966: 284–289. doi: 10.1111/j.1749-6632.2002.tb04228.x
|
| [140] |
Cutolo M, Maestroni GJ, Otsa K, et al. (2005) Circadian melatonin and cortisol levels in rheumatoid arthritis patients in winter time: a north and south Europe comparison. Ann Rheum Dis 64: 212–216. doi: 10.1136/ard.2004.023416
|
| [141] |
Arkema E V, Hart JE, Bertrand KA, et al. (2013) Exposure to ultraviolet-B and risk of developing rheumatoid arthritis among women in the Nurses’ Health Study. Ann Rheum Dis 72: 506–511. doi: 10.1136/annrheumdis-2012-202302
|
| [142] |
Brainard GC, Podolin PL, Leivy SW, et al. (1986) Near-ultraviolet radiation suppresses pineal melatonin content. Endocrinology 119: 2201–2205. doi: 10.1210/endo-119-5-2201
|
| [143] |
Holick MF (2007) Vitamin D deficiency. N Engl J Med 357: 266–281. doi: 10.1056/NEJMra070553
|
| [144] |
Becklund BR, Severson KS, Vang S V, et al. (2010) UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc Natl Acad Sci U S A 107: 6418–6423. doi: 10.1073/pnas.1001119107
|
| [145] |
Skobowiat C, Slominski AT (2015) UVB Activates Hypothalamic-Pituitary-Adrenal Axis in C57BL/6 Mice. J Invest Dermatol 135: 1638–1648. doi: 10.1038/jid.2014.450
|
| [146] |
Cutolo M, Masi AT (2005) Circadian rhythms and arthritis. Rheum Dis Clin North Am 31: 115–129. doi: 10.1016/j.rdc.2004.09.005
|
| [147] |
Cutolo M (2012) Chronobiology and the treatment of rheumatoid arthritis. Curr Opin Rheumatol 24: 312–318. doi: 10.1097/BOR.0b013e3283521c78
|
| [148] |
Petrovsky N, Harrison LC (1998) The chronobiology of human cytokine production. Int Rev Immunol 16: 635–649. doi: 10.3109/08830189809043012
|
| [149] |
Cutolo M, Straub RH (2008) Circadian rhythms in arthritis: hormonal effects on the immune/inflammatory reaction. Autoimmun Rev 7: 223–228. doi: 10.1016/j.autrev.2007.11.019
|
| [150] |
El-Awady HM, El-Wakkad AS, Saleh MT, et al. (2007) Serum melatonin in juvenile rheumatoid arthritis: correlation with disease activity. Pak J Biol Sci 10: 1471–1476. doi: 10.3923/pjbs.2007.1471.1476
|
| [151] |
Afkhamizadeh M, Sahebari M, Seyyed-Hoseini SR (2014) Morning melatonin serum values do not correlate with disease activity in rheumatoid arthritis: a cross-sectional study. Rheumatol Int 34: 1145–1151. doi: 10.1007/s00296-013-2930-x
|
| [152] | West SK, Oosthuizen JM (1992) Melatonin levels are decreased in rheumatoid arthritis. J Basic Clin Physiol Pharmacol 3: 33–40. |
| [153] |
Sulli A, Maestroni GJ, Villaggio B, et al. (2002) Melatonin serum levels in rheumatoid arthritis. Ann N Y Acad Sci 966: 276–283. doi: 10.1111/j.1749-6632.2002.tb04227.x
|
| [154] |
Forrest CM, Mackay GM, Stoy N, et al. (2007) Inflammatory status and kynurenine metabolism in rheumatoid arthritis treated with melatonin. Br J Clin Pharmacol 64: 517–526. doi: 10.1111/j.1365-2125.2007.02911.x
|
| [155] |
Maestroni GJ, Sulli A, Pizzorni C, et al. (2002) Melatonin in rheumatoid arthritis: synovial macrophages show melatonin receptors. Ann N Y Acad Sci 966: 271–275. doi: 10.1111/j.1749-6632.2002.tb04226.x
|
| [156] |
Cutolo M, Villaggio B, Candido F, et al. (1999) Melatonin influences interleukin-12 and nitric oxide production by primary cultures of rheumatoid synovial macrophages and THP-1 cells. Ann N Y Acad Sci 876: 246–254. doi: 10.1111/j.1749-6632.1999.tb07645.x
|
| [157] |
Kouri VP, Olkkonen J, Kaivosoja E, et al. (2013) Circadian timekeeping is disturbed in rheumatoid arthritis at molecular level. PLoS One 8: 54049–54059. doi: 10.1371/journal.pone.0054049
|
| [158] |
Nah SS, Won HJ, Park HJ, et al. (2009) Melatonin inhibits human fibroblast-like synoviocyte proliferation via extracellular signal-regulated protein kinase/P21(CIP1)/P27(KIP1) pathways. J Pineal Res 47: 70–74. doi: 10.1111/j.1600-079X.2009.00689.x
|
| [159] |
Liu X, Xu Y, Chen S, et al. (2014) Rescue of proinflammatory cytokine-inhibited chondrogenesis by the antiarthritic effect of melatonin in synovium mesenchymal stem cells via suppression of reactive oxygen species and matrix metalloproteinases. Free Radic Biol Med 68: 234–246. doi: 10.1016/j.freeradbiomed.2013.12.012
|
| [160] |
Lin GJ, Huang SH, Chen SJ, et al. (2013) Modulation by melatonin of the pathogenesis of inflammatory autoimmune diseases. Int J Mol Sci 14: 11742–11766. doi: 10.3390/ijms140611742
|
| [161] | Arushanian EB, Naumov SS, Ivanova VN, et al. (2014) Comparative study of the influence of melatonin and diclofenac on some hematologic indices of experimental rheumatoid arthritis in rats. Eksp Klin Farmakol 77: 13–15. |
| [162] |
Steinman L (1996) Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85: 299–302. doi: 10.1016/S0092-8674(00)81107-1
|
| [163] |
Pugliatti M, Sotgiu S, Rosati G (2002) The worldwide prevalence of multiple sclerosis. Clin Neurol Neurosurg 104: 182–191. doi: 10.1016/S0303-8467(02)00036-7
|
| [164] |
Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis-the plaque and its pathogenesis. N Engl J Med 354: 942–955. doi: 10.1056/NEJMra052130
|
| [165] |
Lassmann H, van HJ (2011) The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett 585: 3715–3723. doi: 10.1016/j.febslet.2011.08.004
|
| [166] |
Glabinski A, Tawsek NS, Bartosz G (1993) Increased generation of superoxide radicals in the blood of MS patients. Acta Neurol Scand 88: 174–177. doi: 10.1111/j.1600-0447.1993.tb03434.x
|
| [167] |
Besler HT, Comoglu S (2003) Lipoprotein oxidation, plasma total antioxidant capacity and homocysteine level in patients with multiple sclerosis. Nutr Neurosci 6: 189–196. doi: 10.1080/1028415031000115945
|
| [168] |
MacMicking JD, Willenborg DO, Weidemann MJ, et al. (1992) Elevated secretion of reactive nitrogen and oxygen intermediates by inflammatory leukocytes in hyperacute experimental autoimmune encephalomyelitis: enhancement by the soluble products of encephalitogenic T cells. J Exp Med 176: 303–307. doi: 10.1084/jem.176.1.303
|
| [169] |
Hutter CD, Laing P (1996) Multiple sclerosis: sunlight, diet, immunology and aetiology. Med Hypotheses 46: 67–74. doi: 10.1016/S0306-9877(96)90002-X
|
| [170] |
Van der Mei IA, Ponsonby AL, Dwyer T, et al. (2003) Past exposure to sun, skin phenotype, and risk of multiple sclerosis: case-control study. BMJ 327: 316–322. doi: 10.1136/bmj.327.7410.316
|
| [171] |
Islam T, Gauderman WJ, Cozen W, et al. (2007) Childhood sun exposure influences risk of multiple sclerosis in monozygotic twins. Neurology 69: 381–388. doi: 10.1212/01.wnl.0000268266.50850.48
|
| [172] |
Mehta BK (2010) New hypotheses on sunlight and the geographic variability of multiple sclerosis prevalence. J Neurol Sci 292: 5–10. doi: 10.1016/j.jns.2010.02.004
|
| [173] | Ghorbani A, Salari M, Shaygannejad V, et al. (2013) The role of melatonin in the pathogenesis of multiple sclerosis: a case-control study. Int J Prev Med 4: S180–S184. |
| [174] |
Kurtzke JF (1977) Geography in multiple sclerosis. J Neurol 215: 1–26. doi: 10.1007/BF00312546
|
| [175] |
Kurtzke JF (1967) On the fine structure of the distribution of multiple sclerosis. Acta Neurol Scand 43: 257–282. doi: 10.1111/j.1600-0404.1967.tb05733.x
|
| [176] |
Natarajan R, Einarsdottir E, Riutta A, et al. (2012) Melatonin pathway genes are associated with progressive subtypes and disability status in multiple sclerosis among Finnish patients. J Neuroimmunol 250: 106–110. doi: 10.1016/j.jneuroim.2012.05.014
|
| [177] |
Sandyk R (1993) Multiple sclerosis: the role of puberty and the pineal gland in its pathogenesis. Int J Neurosci 68: 209–225. doi: 10.3109/00207459308994277
|
| [178] |
Hedstrom AK, Akerstedt T, Hillert J, et al. (2011) Shift work at young age is associated with increased risk for multiple sclerosis. Ann Neurol 70: 733–741. doi: 10.1002/ana.22597
|
| [179] |
Brass SD, Duquette P, Proulx-Therrien J, et al. (2010) Sleep disorders in patients with multiple sclerosis. Sleep Med Rev 14: 121–129. doi: 10.1016/j.smrv.2009.07.005
|
| [180] | Barun B (2013) Pathophysiological background and clinical characteristics of sleep disorders in multiple sclerosis. Clin Neurol Neurosurg 115 Suppl1: S82–S85. |
| [181] |
Sandyk R, Awerbuch GI (1992) Nocturnal plasma melatonin and alpha-melanocyte stimulating hormone levels during exacerbation of multiple sclerosis. Int J Neurosci 67: 173–186. doi: 10.3109/00207459208994783
|
| [182] | Gholipour T, Ghazizadeh T, Babapour S, et al. (2015) Decreased urinary level of melatonin as a marker of disease severity in patients with multiple sclerosis. Iran J Allergy, Asthma Immunol 14: 91–97. |
| [183] |
Melamud L, Golan D, Luboshitzky R, et al. (2012) Melatonin dysregulation, sleep disturbances and fatigue in multiple sclerosis. J Neurol Sci 314: 37–40. doi: 10.1016/j.jns.2011.11.003
|
| [184] |
Escribano BM, Colin-Gonzalez AL, Santamaria A, et al. (2014) The role of melatonin in multiple sclerosis, Huntington’s disease and cerebral ischemia. CNS Neurol Disord Drug Targets 13: 1096–1119. doi: 10.2174/1871527313666140806160400
|
| [185] |
Damasceno A, Moraes AS, Farias A, et al. (2015) Disruption of melatonin circadian rhythm production is related to multiple sclerosis severity: A preliminary study. J Neurol Sci 353: 166–168. doi: 10.1016/j.jns.2015.03.040
|
| [186] |
Akpinar Z, Tokgoz S, Gokbel H, et al. (2008) The association of nocturnal serum melatonin levels with major depression in patients with acute multiple sclerosis. Psychiatry Res 161: 253–257. doi: 10.1016/j.psychres.2007.11.022
|
| [187] |
Sandyk R (1995) Diurnal variations in vision and relations to circadian melatonin secretion in multiple sclerosis. Int J Neurosci 83: 1–6. doi: 10.3109/00207459508986320
|
| [188] |
Anderson G, Rodriguez M (2011) Multiple sclerosis, seizures, and antiepileptics: role of IL-18, IDO, and melatonin. Eur J Neurol 18: 680–685. doi: 10.1111/j.1468-1331.2010.03257.x
|
| [189] |
Maestroni GJ (2001) The immunotherapeutic potential of melatonin. Expert Opin Investig Drugs 10: 467–476. doi: 10.1517/13543784.10.3.467
|
| [190] | Adamczyk-Sowa M, Pierzchala K, Sowa P, et al. (2014) Influence of melatonin supplementation on serum antioxidative properties and impact of the quality of life in multiple sclerosis patients. J Physiol Pharmacol 65: 543–550. |
| [191] |
Emamgholipour S, Hossein-nezhad A, Sahraian MA, et al. (2016) Evidence for possible role of melatonin in reducing oxidative stress in multiple sclerosis through its effect on SIRT1 and antioxidant enzymes. Life Sci 145: 34–41. doi: 10.1016/j.lfs.2015.12.014
|
| [192] |
López-González A, álvarez-Sánchez N, Lardone PJ, et al. (2015) Melatonin treatment improves primary progressive multiple sclerosis: a case report. J Pineal Res 58: 173–177. doi: 10.1111/jpi.12203
|
| [193] |
Bahamonde C, Conde C, Aguera E, et al. (2014) Elevated melatonin levels in natalizumab-treated female patients with relapsing-remitting multiple sclerosis: relationship to oxidative stress. Eur J Pharmacol 730: 26–30. doi: 10.1016/j.ejphar.2014.02.020
|
| [194] | Kang JC, Ahn M, Kim YS, et al. (2001) Melatonin ameliorates autoimmune encephalomyelitis through suppression of intercellular adhesion molecule-1. J Vet Sci 2: 85–89. |
| [195] |
Kashani IR, Rajabi Z, Akbari M, et al. (2014) Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res 232: 2835–2846. doi: 10.1007/s00221-014-3946-5
|
| [196] |
álvarez-Sánchez N, Cruz-Chamorro I, López-González A, et al. (2015) Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav Immun 50: 101–114. doi: 10.1016/j.bbi.2015.06.021
|
| [197] | Chen S-J, Huang S-H, Chen J-W, et al. (2015) Melatonin enhances interleukin-10 expression and suppresses chemotaxis to inhibit inflammation in situ and reduce the severity of experimental autoimmune encephalomyelitis. Int Imunopharmacology 31: 169–177. |
| [198] |
Farez MF, Mascanfroni ID, Méndez-Huergo SP, et al. (2015) Melatonin Contributes to the Seasonality of Multiple Sclerosis Relapses. Cell 162: 1338–1352. doi: 10.1016/j.cell.2015.08.025
|
| [199] |
Lee JS, Cua DJ (2015) Melatonin lulling Th17 cells to sleep. Cell 162: 1212–1214. doi: 10.1016/j.cell.2015.08.054
|
| [200] |
Constantinescu CS, Hilliard B, Ventura E, et al. (1997) Luzindole, a melatonin receptor antagonist, suppresses experimental autoimmune encephalomyelitis. Pathobiology 65: 190–194. doi: 10.1159/000164122
|
| [201] | Dubocovich ML, Yun K, Al-Ghoul WM, et al. (1998) Selective MT2 melatonin receptor antagonists block melatonin-mediated phase advances of circadian rhythms. FASEB J 12: 1211–1220. |
| [202] |
Sandyk R (1997) Influence of the pineal gland on the expression of experimental allergic encephalomyelitis: possible relationship to the aquisition of multiple sclerosis. Int J Neurosci 90: 129–133. doi: 10.3109/00207459709000632
|
| [203] |
D’Cruz DP, Khamashta MA, Hughes GR (2007) Systemic lupus erythematosus. Lancet 369: 587–596. doi: 10.1016/S0140-6736(07)60279-7
|
| [204] |
Jancar S, Sanchez CM (2005) Immune complex-mediated tissue injury: a multistep paradigm. Trends Immunol 26: 48–55. doi: 10.1016/j.it.2004.11.007
|
| [205] | Kidd P (2003) Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev 8: 223–246. |
| [206] | Hayashi T (2010) Therapeutic strategies for SLE involving cytokines: mechanism-oriented therapies especially IFN-gamma targeting gene therapy. J Biomed Biotechnol 2010: 461641–461660. |
| [207] |
Wong CK, Lit LC, Tam LS, et al. (2008) Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol 127: 385–393. doi: 10.1016/j.clim.2008.01.019
|
| [208] |
Fairhurst AM, Wandstrat AE, Wakeland EK (2006) Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol 92: 1–69. doi: 10.1016/S0065-2776(06)92001-X
|
| [209] |
Tsokos GC (2011) Systemic lupus erythematosus. N Engl J Med 365: 2110–2121. doi: 10.1056/NEJMra1100359
|
| [210] |
Lenz SP, Izui S, Benediktsson H, et al. (1995) Lithium chloride enhances survival of NZB/W lupus mice: influence of melatonin and timing of treatment. Int J Immunopharmacol 17: 581–592. doi: 10.1016/0192-0561(95)00032-W
|
| [211] |
Lechner O, Dietrich H, Oliveira dos SA, et al. (2000) Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J Autoimmun 14: 325–333. doi: 10.1006/jaut.2000.0375
|
| [212] |
Yin Z, Bahtiyar G, Zhang N, et al. (2002) IL-10 regulates murine lupus. J Immunol 169: 2148–2155. doi: 10.4049/jimmunol.169.4.2148
|
| [213] |
Jimenez-Caliani AJ, Jimenez-Jorge S, Molinero P, et al. (2006) Sex-dependent effect of melatonin on systemic erythematosus lupus developed in Mrl/Mpj-Faslpr mice: it ameliorates the disease course in females, whereas it exacerbates it in males. Endocrinology 147: 1717–1724. doi: 10.1210/en.2005-0648
|
| [214] |
Jimenez-Caliani AJ, Jimenez-Jorge S, Molinero P, et al. (2008) Treatment with testosterone or estradiol in melatonin treated females and males MRL/MpJ-Faslpr mice induces negative effects in developing systemic lupus erythematosus. J Pineal Res 45: 204–211. doi: 10.1111/j.1600-079X.2008.00578.x
|
| [215] | Zhou LL, Wei W, Si JF, et al. (2010) Regulatory effect of melatonin on cytokine disturbances in the pristane-induced lupus mice. Mediat Inflamm 2010: 951210–951217. |
| [216] |
Wu CC, Lu KC, Lin GJ, et al. (2012) Melatonin enhances endogenous heme oxygenase-1 and represses immune responses to ameliorate experimental murine membranous nephropathy. J Pineal Res 52: 460–469. doi: 10.1111/j.1600-079X.2011.00960.x
|
| [217] |
Haga HJ, Brun JG, Rekvig OP, et al. (1999) Seasonal variations in activity of systemic lupus erythematosus in a subarctic region. Lupus 8: 269–273. doi: 10.1191/096120399678847858
|
| [218] | Robeva R, Tanev D, Kirilov G, et al. (2013) Decreased daily melatonin levels in women with systemic lupus erythematosus - a short report. Balk Med J 30: 273–276. |
| [219] | Kawasaki E, Abiru N, Eguchi K (2004) Prevention of type 1 diabetes: from the view point of beta cell damage. Diabetes Res Clin Pr 66 Suppl 1: S27–S32. |
| [220] |
Daneman D (2006) Type 1 diabetes. Lancet 367: 847–858. doi: 10.1016/S0140-6736(06)68341-4
|
| [221] |
Ponsonby AL, Lucas RM, van Ider M (2005) UVR, vitamin D and three autoimmune diseases--multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol 81: 1267–1275. doi: 10.1562/2005-02-15-IR-441
|
| [222] |
Aoki CA, Borchers AT, Ridgway WM, et al. (2005) NOD mice and autoimmunity. Autoimmun Rev 4: 373–379. doi: 10.1016/j.autrev.2005.02.002
|
| [223] |
Wicker LS, Todd JA, Peterson LB (1995) Genetic control of autoimmune diabetes in the NOD mouse. Annu Rev Immunol 13: 179–200. doi: 10.1146/annurev.iy.13.040195.001143
|
| [224] |
Liblau RS, Singer SM, McDevitt HO (1995) Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today 16: 34–38. doi: 10.1016/0167-5699(95)80068-9
|
| [225] |
Hung JT, Liao JH, Lin YC, et al. (2005) Immunopathogenic role of TH1 cells in autoimmune diabetes: evidence from a T1 and T2 doubly transgenic non-obese diabetic mouse model. J Autoimmun 25: 181–192. doi: 10.1016/j.jaut.2005.08.010
|
| [226] |
Koarada S, Wu Y, Olshansky G, et al. (2002) Increased nonobese diabetic Th1: Th2 (IFN-gamma: IL-4) ratio is CD4+ T cell intrinsic and independent of APC genetic background. J Immunol 169: 6580–6587. doi: 10.4049/jimmunol.169.11.6580
|
| [227] |
Conti A, Maestroni GJ (1996) Role of the pineal gland and melatonin in the development of autoimmune diabetes in non-obese diabetic mice. J Pineal Res 20: 164–172. doi: 10.1111/j.1600-079X.1996.tb00253.x
|
| [228] |
Lin GJ, Huang SH, Chen YW, et al. (2009) Melatonin prolongs islet graft survival in diabetic NOD mice. J Pineal Res 47: 284–292. doi: 10.1111/j.1600-079X.2009.00712.x
|
| [229] |
Peschke E, Wolgast S, Bazwinsky I, et al. (2008) Increased melatonin synthesis in pineal glands of rats in streptozotocin induced type 1 diabetes. J Pineal Res 45: 439–448. doi: 10.1111/j.1600-079X.2008.00612.x
|
| [230] | Peschke E, Hofmann K, Bahr I, et al. (2011) The insulin-melatonin antagonism: studies in the LEW.1AR1-iddm rat (an animal model of human type 1 diabetes mellitus). Diabetologia 54: 1831–1840. |
| [231] |
Peschke E, Hofmann K, Ponicke K, et al. (2012) Catecholamines are the key for explaining the biological relevance of insulin-melatonin antagonisms in type 1 and type 2 diabetes. J Pineal Res 52: 389–396. doi: 10.1111/j.1600-079X.2011.00951.x
|
| [232] |
Kor Y, Geyikli I, Keskin M, et al. (2014) Preliminary study: Evaluation of melatonin secretion in children and adolescents with type 1 diabetes mellitus. Indian J Endocrinol Metab 18: 565–568. doi: 10.4103/2230-8210.137521
|
| [233] |
Reyes-Toso CF, Roson MI, Albornoz LE, et al. (2002) Vascular reactivity in diabetic rats: effect of melatonin. J Pineal Res 33: 81–86. doi: 10.1034/j.1600-079X.2002.01886.x
|
| [234] |
Cavallo A, Daniels SR, Dolan LM, et al. (2004) Blood pressure-lowering effect of melatonin in type 1 diabetes. J Pineal Res 36: 262–266. doi: 10.1111/j.1600-079X.2004.00126.x
|
| [235] | Abraham C, Cho JH (2009) Inflammatory bowel disease. N Engl J Med 19: 2066–2078. |
| [236] |
Motilva V, Garcia-Maurino S, Talero E, et al. (2011) New paradigms in chronic intestinal inflammation and colon cancer: role of melatonin. J Pineal Res 51: 44–60. doi: 10.1111/j.1600-079X.2011.00915.x
|
| [237] |
Sartor RB (2006) Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pr Gastroenterol Hepatol 3: 390–407. doi: 10.1038/ncpgasthep0528
|
| [238] |
Neurath MF, Finotto S (2006) The many roads to inflammatory bowel diseases. Immunity 25: 189–191. doi: 10.1016/j.immuni.2006.08.005
|
| [239] | Fuss IJ, Neurath M, Boirivant M, et al. (1996) Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 157: 1261–1270. |
| [240] |
Fujino S, Andoh A, Bamba S, et al. (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52: 65–70. doi: 10.1136/gut.52.1.65
|
| [241] |
Hue S, Ahern P, Buonocore S, et al. (2006) Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med 203: 2473–2483. doi: 10.1084/jem.20061099
|
| [242] |
Elson CO, Cong Y, Weaver CT, et al. (2007) Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132: 2359–2370. doi: 10.1053/j.gastro.2007.03.104
|
| [243] |
Barrett JC, Hansoul S, Nicolae DL, et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40: 955–962. doi: 10.1038/ng.175
|
| [244] |
Preuss F, Tang Y, Laposky AD, et al. (2008) Adverse effects of chronic circadian desynchronization in animals in a “challenging” environment. Am J Physiol Regul Integr Comp Physiol 295: R2034–R2040. doi: 10.1152/ajpregu.00118.2008
|
| [245] |
Tang Y, Preuss F, Turek FW, et al. (2009) Sleep deprivation worsens inflammation and delays recovery in a mouse model of colitis. Sleep Med 10: 597–603. doi: 10.1016/j.sleep.2008.12.009
|
| [246] |
Mickle A, Sood M, Zhang Z, et al. (2010) Antinociceptive effects of melatonin in a rat model of post-inflammatory visceral hyperalgesia: a centrally mediated process. Pain 149: 555–564. doi: 10.1016/j.pain.2010.03.030
|
| [247] |
Pentney PT, Bubenik GA (1995) Melatonin reduces the severity of dextran-induced colitis in mice. J Pineal Res 19: 31–39. doi: 10.1111/j.1600-079X.1995.tb00168.x
|
| [248] |
Cuzzocrea S, Mazzon E, Serraino I, et al. (2001) Melatonin reduces dinitrobenzene sulfonic acid-induced colitis. J Pineal Res 30: 1–12. doi: 10.1034/j.1600-079X.2001.300101.x
|
| [249] |
Trivedi PP, Jena GB (2013) Melatonin reduces ulcerative colitis-associated local and systemic damage in mice: investigation on possible mechanisms. Dig Dis Sci 58: 3460–3474. doi: 10.1007/s10620-013-2831-6
|
| [250] | Park Y-S, Chung S-H, Lee S-K, et al. (2015) Melatonin improves experimental colitis with sleep deprivation. Int J Mol Med 35: 979–986. |
| [251] |
Dong WG, Mei Q, Yu JP, et al. (2003) Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J Gastroenterol 9: 1307–1311. doi: 10.3748/wjg.v9.i6.1307
|
| [252] |
Tahan G, Gramignoli R, Marongiu F, et al. (2011) Melatonin expresses powerful anti-inflammatory and antioxidant activities resulting in complete improvement of acetic-acid-induced colitis in rats. Dig Dis Sci 56: 715–720. doi: 10.1007/s10620-010-1364-5
|
| [253] |
Akcan A, Kucuk C, Sozuer E, et al. (2008) Melatonin reduces bacterial translocation and apoptosis in trinitrobenzene sulphonic acid-induced colitis of rats. World J Gastroenterol 14: 918–924. doi: 10.3748/wjg.14.918
|
| [254] |
Esposito E, Mazzon E, Riccardi L, et al. (2008) Matrix metalloproteinase-9 and metalloproteinase-2 activity and expression is reduced by melatonin during experimental colitis. J Pineal Res 45: 166–173. doi: 10.1111/j.1600-079X.2008.00572.x
|
| [255] |
Mazzon E, Esposito E, Crisafulli C, et al. (2006) Melatonin modulates signal transduction pathways and apoptosis in experimental colitis. J Pineal Res 41: 363–373. doi: 10.1111/j.1600-079X.2006.00378.x
|
| [256] |
Li JH, Yu JP, Yu HG, et al. (2005) Melatonin reduces inflammatory injury through inhibiting NF-kappaB activation in rats with colitis. Mediat Inflamm 2005: 185–193. doi: 10.1155/MI.2005.185
|
| [257] |
Tasdemir S, Parlakpinar H, Vardi N, et al. (2013) Effect of endogen-exogenous melatonin and erythropoietin on dinitrobenzene sulfonic acid-induced colitis. Fundam Clin Pharmacol 27: 299–307. doi: 10.1111/j.1472-8206.2011.01016.x
|
| [258] |
Mann S (2003) Melatonin for ulcerative colitis? Am J Gastroenterol 98: 232–233. doi: 10.1111/j.1572-0241.2003.07190.x
|
| [259] |
Calvo JR, Guerrero JM, Osuna C, et al. (2002) Melatonin triggers Crohn’s disease symptoms. J Pineal Res 32: 277–278. doi: 10.1034/k.1600-079X.2002.01881.x
|
| [260] |
Maldonado MD, Calvo JR (2008) Melatonin usage in ulcerative colitis: a case report. J Pineal Res 45: 339–340. doi: 10.1111/j.1600-079X.2008.00584.x
|
| [261] | Rakhimova OI (2010) Use of melatonin in combined treatment for inflammatory bowel diseases. Ter Arkh 82: 64–68. |
| [262] |
Mozaffari S, Abdollahi M (2011) Melatonin, a promising supplement in inflammatory bowel disease: a comprehensive review of evidences. Curr Pharm Des 17: 4372–4378. doi: 10.2174/138161211798999357
|
| [263] |
Jena G, Trivedi PP (2014) A review of the use of melatonin in ulcerative colitis: experimental evidence and new approaches. Inflamm Bowel Dis 20: 553–563. doi: 10.1097/01.MIB.0000436962.32164.6e
|
| [264] | Chojnacki C, Wisniewska-Jarosinska M, Walecka-Kapica E, et al. (2011) Evaluation of melatonin effectiveness in the adjuvant treatment of ulcerative colitis. J Physiol Pharmacol 62: 327–334. |
| [265] |
Yang J, Carra S, Zhu WG, et al. (2013) The regulation of the autophagic network and its implications for human disease. Int J Biol Sci 9: 1121–1133. doi: 10.7150/ijbs.6666
|
| [266] |
Cheng Y, Ren X, Hait WN, et al. (2013) Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 65: 1162–1197. doi: 10.1124/pr.112.007120
|
| [267] | Ryter SW, Mizumura K, Choi AM (2014) The impact of autophagy on cell death modalities. Int J Cell Biol 2014: 502676–502688. |
| [268] |
Talero E, Garcia-Maurino S, Motilva V (2014) Melatonin, autophagy and intestinal bowel disease. Curr Pharm Des 20: 4816–4827. doi: 10.2174/1381612819666131119110835
|
| [269] |
Swanson GR, Burgess HJ, Keshavarzian A (2011) Sleep disturbances and inflammatory bowel disease: a potential trigger for disease flare? Expert Rev Clin Immunol 7: 29–36. doi: 10.1586/eci.10.83
|
| [270] | Takagi T, Inada Y, Naito Y (2013) Circadian rhythm and inflammatory bowel disease. Nihon Rinsho 71: 2165–2170. |
| [271] |
Chung SH, Park YS, Kim OS, et al. (2014) Melatonin attenuates dextran sodium sulfate induced colitis with sleep deprivation: possible mechanism by microarray analysis. Dig Dis Sci 59: 1134–1141. doi: 10.1007/s10620-013-3013-2
|
| [272] |
Anderson G, Rodriguez M (2015) Multiple sclerosis: the role of melatonin and N-acetylserotonin. Mult Scler Relat Disord 4: 112–123. doi: 10.1016/j.msard.2014.12.001
|
| [273] | Miller E, Morel A, Saso L, et al. (2015) Melatonin redox activity. Its potential clinical applications in neurodegenerative disorders. Curr Top Med Chem 15: 163–169. |
| [274] |
Carrillo-Vico A, Guerrero JM, Lardone PJ, et al. (2005) A review of the multiple actions of melatonin on the immune system. Endocrine 27: 189–200. doi: 10.1385/ENDO:27:2:189
|
| [275] |
Pandi-Perumal SR, Srinivasan V, Maestroni GJM, et al. (2006) Melatonin: Nature’s most versatile biological signal? FEBS J 273: 2813–2838. doi: 10.1111/j.1742-4658.2006.05322.x
|
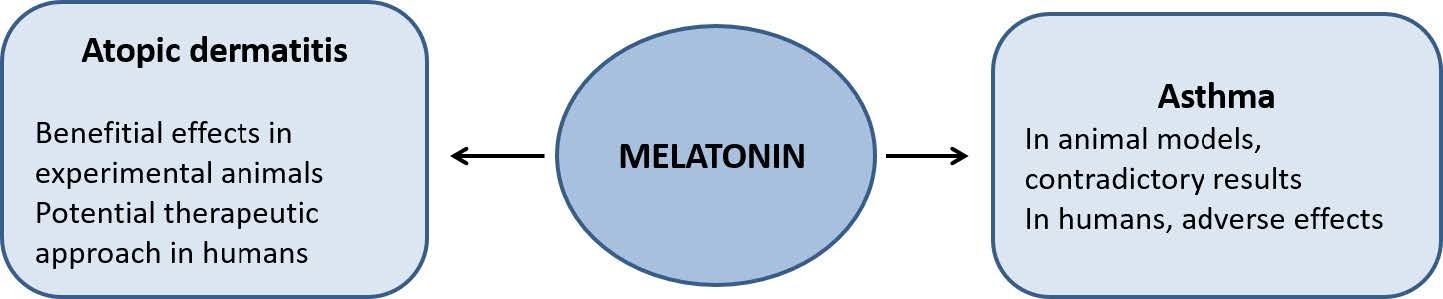
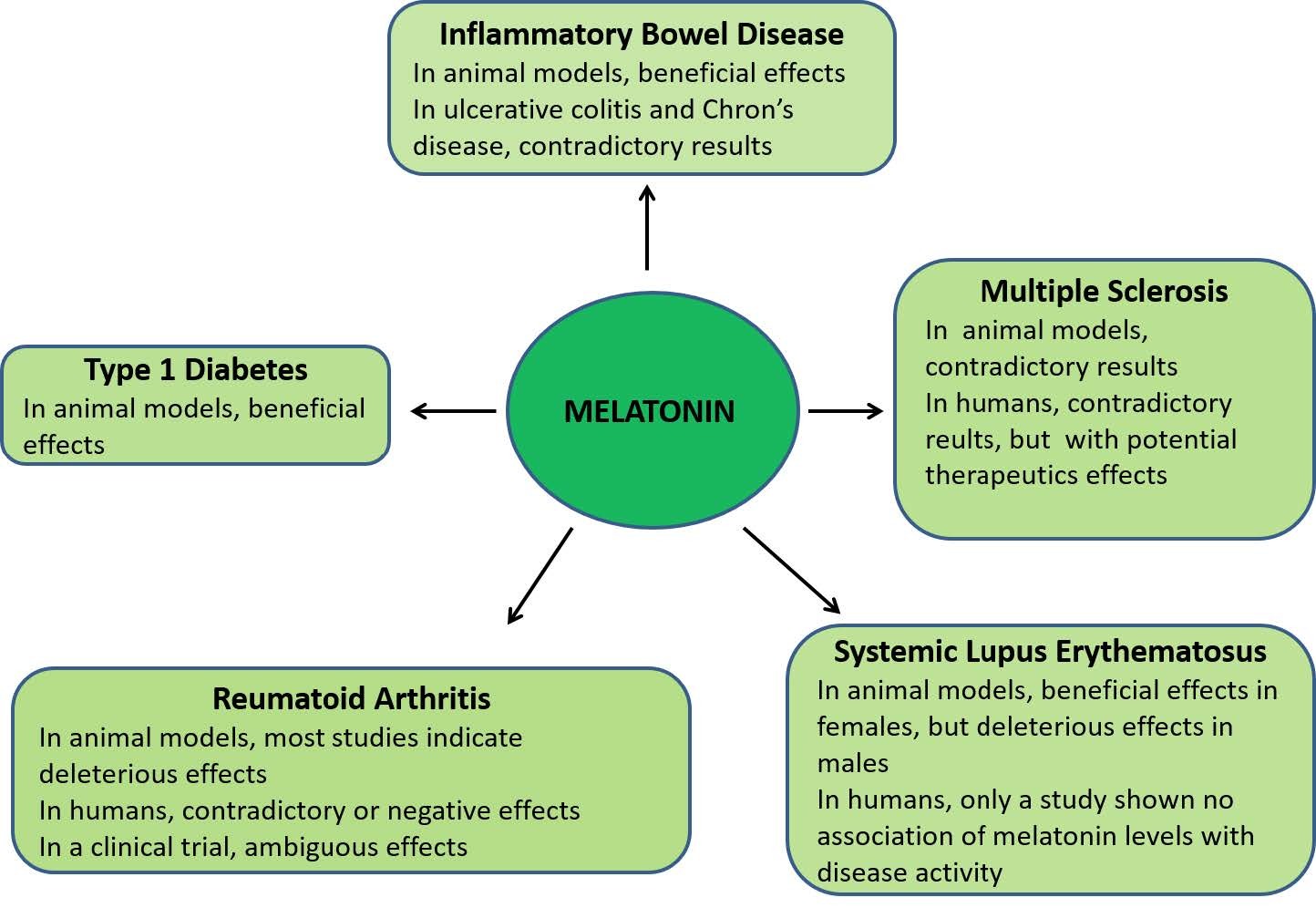
Figures(2)

J.R. Calvo, M.D. Maldonado. The role of melatonin in autoimmune and atopic diseases[J]. AIMS Molecular Science, 2016, 3(2): 158-186. doi: 10.3934/molsci.2016.2.158







 DownLoad:
DownLoad: