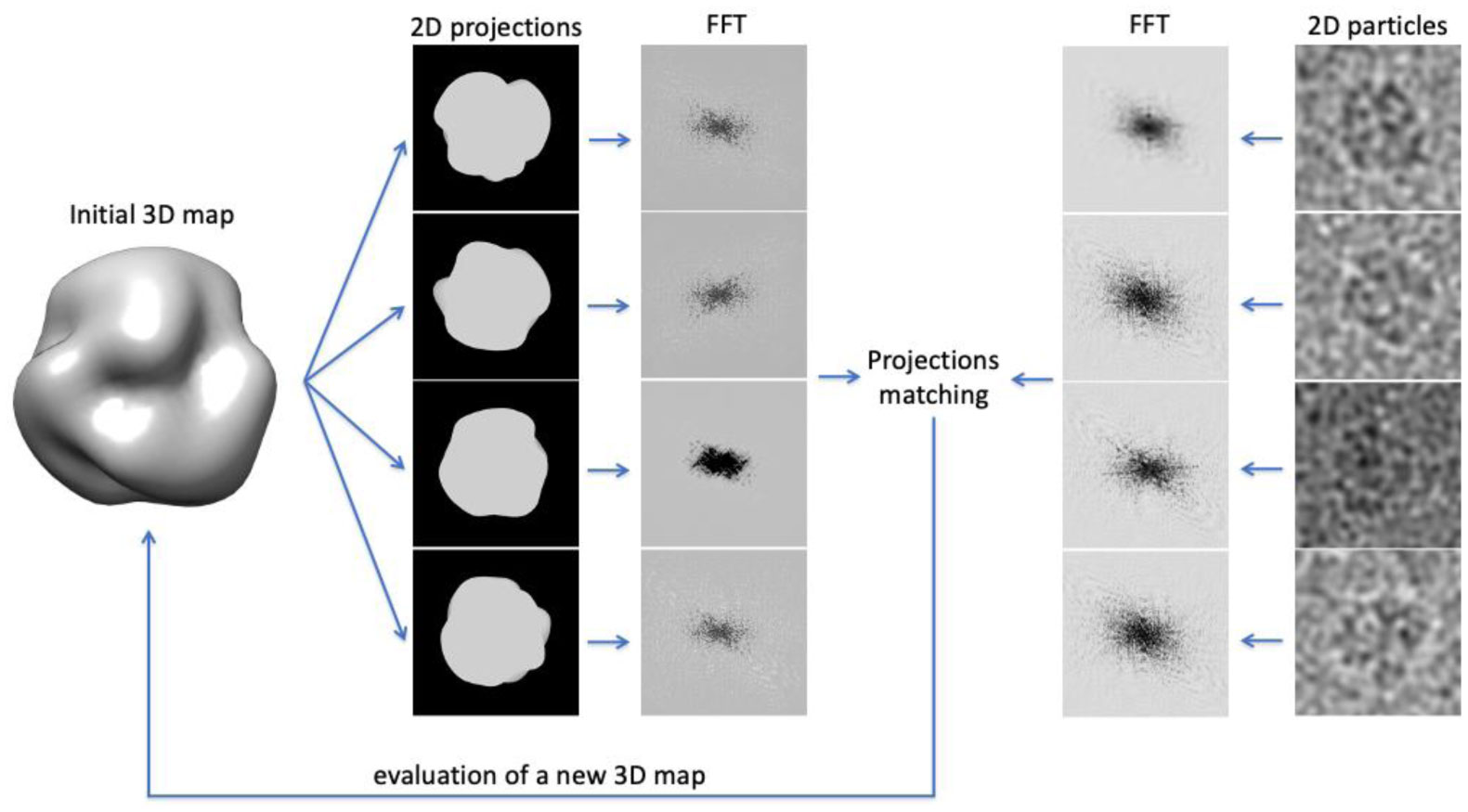
In the last years, cryogenic-electron microscopy (cryo-EM) underwent the most impressive improvement compared to other techniques used in structural biology, such as X-ray crystallography and NMR. Electron microscopy was invented nearly one century ago but, up to the beginning of the last decades, the 3D maps produced through this technique were poorly detailed, justifying the term “blobbology” to appeal to cryo-EM. Recently, thanks to a new generation of microscopes and detectors, more efficient algorithms, and easier access to computational power, single particles cryo-EM can routinely produce 3D structures at resolutions comparable to those obtained with X-ray crystallography. However, unlike X-ray crystallography, which needs crystallized proteins, cryo-EM exploits purified samples in solution, allowing the study of proteins and protein complexes that are hard or even impossible to crystallize. For these reasons, single-particle cryo-EM is often the first choice of structural biologists today. Nevertheless, before starting a cryo-EM experiment, many drawbacks and limitations must be considered. Moreover, in practice, the process between the purified sample and the final structure could be trickier than initially expected. Based on these observations, this review aims to offer an overview of the principal technical aspects and setups to be considered while planning and performing a cryo-EM experiment.
Citation: Vittoria Raimondi, Alessandro Grinzato. A basic introduction to single particles cryo-electron microscopy[J]. AIMS Biophysics, 2022, 9(1): 5-20. doi: 10.3934/biophy.2022002
In the last years, cryogenic-electron microscopy (cryo-EM) underwent the most impressive improvement compared to other techniques used in structural biology, such as X-ray crystallography and NMR. Electron microscopy was invented nearly one century ago but, up to the beginning of the last decades, the 3D maps produced through this technique were poorly detailed, justifying the term “blobbology” to appeal to cryo-EM. Recently, thanks to a new generation of microscopes and detectors, more efficient algorithms, and easier access to computational power, single particles cryo-EM can routinely produce 3D structures at resolutions comparable to those obtained with X-ray crystallography. However, unlike X-ray crystallography, which needs crystallized proteins, cryo-EM exploits purified samples in solution, allowing the study of proteins and protein complexes that are hard or even impossible to crystallize. For these reasons, single-particle cryo-EM is often the first choice of structural biologists today. Nevertheless, before starting a cryo-EM experiment, many drawbacks and limitations must be considered. Moreover, in practice, the process between the purified sample and the final structure could be trickier than initially expected. Based on these observations, this review aims to offer an overview of the principal technical aspects and setups to be considered while planning and performing a cryo-EM experiment.

| [1] |
Murata K, Wolf M (2018) Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim Biophys Acta-Gen Subj 1862: 324-334. https://doi.org/10.1016/j.bbagen.2017.07.020 
|
| [2] |
Zanotti G, Grinzato A (2021) Structure of filamentous viruses. Curr Opin Virol 51: 25-33. https://doi.org/10.1016/j.coviro.2021.09.006
|
| [3] |
Ruska E (1987) The development of the electron microscope and of electron microscopy. Biosci Rep 7: 607-629. https://doi.org/10.1007/BF01127674
|
| [4] |
Brenner S, Horne RW (1959) A negative staining method for high resolution electron microscopy of viruses. Biochim Biophys Acta 34: 103-110. https://doi.org/10.1016/0006-3002(59)90237-9
|
| [5] |
Von Borries B, Ruska E, Ruska H (1938) Bakterien und virus in übermikroskopischer aufnahme. Klin Wochenschr 17: 921-925. https://doi.org/10.1007/BF01775798
|
| [6] |
Kausche GA, Pfankuch E, Ruska H (1939) Die sichtbarmachung von pflanzlichem virus im Übermikroskop. Naturwissenschaften 27: 292-299. https://doi.org/10.1007/BF01493353
|
| [7] |
Dubochet J, McDowall AW (1981) Vitrification of pure water for electron microscopy. J Microsc 124: 3-4.
|
| [8] |
Dubochet J, Lepault J, Freeman R, et al. (1982) Electron microscopy of frozen water and aqueous solutions. J Microsc 128: 219-237. https://doi.org/10.1111/j.1365-2818.1982.tb04625.x
|
| [9] |
Dubochet J, Adrian M, Chang JJ, et al. (1988) Cryoelectron microscopy of vitrified specimens. Q Rev Biophys 21: 129-228.
|
| [10] |
Frank J, Shimkin B, Dowse H (1981) SPIDER—a modular software system for electron image processing. Ultramicroscopy 6: 343-357. https://doi.org/10.1142/9789813234864_0008
|
| [11] | Van Heel M, Frank J (1981) Use of multivariates statistics in analysing the images of biological macromolecules. Ultramicroscopy 6: 187-194. https://doi.org/10.1016/S0304-3991(81)80197-0 |
| [12] |
Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82-97. https://doi.org/10.1006/jsbi.1999.4174
|
| [13] |
Sorzano COS, Marabini R, Velázquez-Muriel J, et al. (2004) XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol 148: 194-204. https://doi.org/10.1016/j.jsb.2004.06.006
|
| [14] |
Suloway C, Pulokas J, Fellmann D, et al. (2005) Automated molecular microscopy: the new Leginon system. J Struct Biol 151: 41-60. https://doi.org/10.1016/j.jsb.2005.03.010
|
| [15] |
Tang G, Peng L, Baldwin PR, et al. (2007) EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157: 38-46. https://doi.org/10.1016/j.jsb.2006.05.009
|
| [16] |
Lander GC, Stagg SM, Voss NR, et al. (2009) Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166: 95-102. https://doi.org/10.1016/j.jsb.2009.01.002
|
| [17] |
Scheres SHW (2012) RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180: 519-530. https://doi.org/10.1016/j.jsb.2012.09.006
|
| [18] |
Grigorieff N, Grant T, Rohou A (2017) cisTEM: user-friendly software for single-particle image processing. Acta Crystallogr Sect A 73: C1368-C1368.
|
| [19] |
Punjani A, Rubinstein JL, Fleet DJ, et al. (2017) CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14: 290-296. https://doi.org/10.1038/nmeth.4169
|
| [20] |
Conesa Mingo P, Gutierrez J, Quintana A, et al. (2018) Scipion web tools: Easy to use cryo-EM image processing over the web. Protein Sci 27: 269-275. https://doi.org/10.1002/pro.3315
|
| [21] |
Kimanius D, Forsberg BO, Scheres SHW, et al. (2016) Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5: 1-21. https://doi.org/10.7554/eLife.18722.001
|
| [22] | Luecken U, Van HG, Schuurmans F, et al. Method of using a direct electron detector for a TEM (2011). |
| [23] |
Milazzo AC, Cheng A, Moeller A, et al. (2011) Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy. J Struct Biol 176: 404-408. https://doi.org/10.1016/j.jsb.2011.09.002
|
| [24] |
Ruskin RS, Yu Z, Grigorieff N (2013) Quantitative characterization of electron detectors for transmission electron microscopy. J Struct Biol 184: 385-393. https://doi.org/10.1016/j.jsb.2013.10.016
|
| [25] |
Kuijper M, van Hoften G, Janssen B, et al. (2015) FEI's direct electron detector developments: Embarking on a revolution in cryo-TEM. J Struct Biol 192: 179-187. https://doi.org/10.1016/j.jsb.2015.09.014
|
| [26] |
Ripstein ZA, Rubinstein JL (2016) Processing of Cryo-EM movie data. Methods in Enzymology : 103-124. https://doi.org/10.1016/bs.mie.2016.04.009
|
| [27] |
Li X, Mooney P, Zheng S, et al. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10: 584-590. https://doi.org/10.1038/nmeth.2472
|
| [28] |
Kühlbrandt W (2014) The resolution revolution. Science 343: 1443-1444. https://doi.org/10.1126/science.1251652
|
| [29] |
Bai XC, McMullan G, Scheres SHW (2015) How cryo-EM is revolutionizing structural biology. Trends Biochem Sci 40: 49-57. https://doi.org/10.1016/j.tibs.2014.10.005
|
| [30] |
Lawson CL, Patwardhan A, Baker ML, et al. (2016) EMDataBank unified data resource for 3DEM. Nucleic Acids Res 44: D396-403. https://doi.org/10.1093/nar/gkv1126
|
| [31] |
Nakane T, Kotecha A, Sente A, et al. (2020) Single-particle cryo-EM at atomic resolution. Nature 587: 152-156. https://doi.org/10.1038/s41586-020-2829-0
|
| [32] |
Weis F, Beckers M, von der Hocht I, et al. (2019) Elucidation of the viral disassembly switch of tobacco mosaic virus. EMBO Rep 20: e48451. https://doi.org/10.15252/embr.201948451
|
| [33] |
Grinzato A, Kandiah E, Lico C, et al. (2020) Atomic structure of potato virus X, the prototype of the Alphaflexiviridae family. Nat Chem Biol 16: 564-569. https://doi.org/10.1038/s41589-020-0502-4
|
| [34] |
Masuyer G, Conrad J, Stenmark P (2017) The structure of the tetanus toxin reveals pH-mediated domain dynamics. EMBO Rep 18: 1306-1317. https://doi.org/10.15252/embr.201744198
|
| [35] |
Pirazzini M, Grinzato A, Corti D, et al. (2021) Exceptionally potent human monoclonal antibodies are effective for prophylaxis and therapy of tetanus in mice. J Clin Invest 131: e151676. https://doi.org/10.1172/JCI151676
|
| [36] |
Sobti M, Smits C, Wong ASW, et al. (2016) Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. Elife 5: e21598. https://doi.org/10.7554/eLife.21598.001
|
| [37] |
Skiniotis G, Southworth DR (2016) Single-particle cryo-electron microscopy of macromolecular complexes. Microscopy 65: 9-22. https://doi.org/10.1093/jmicro/dfv366
|
| [38] |
Chen JZ, Sachse C, Xu C, et al. (2008) A dose-rate effect in single-particle electron microscopy. J Struct Biol 161: 92-100. https://doi.org/10.1016/j.jsb.2007.09.017
|
| [39] |
Cho H, Hyun J, Kim J, et al. (2013) Measurement of ice thickness on vitreous ice embedded cryo-EM grids: investigation of optimizing condition for visualizing macromolecules. J Anal Sci Technol 4: 7. https://doi.org/10.1186/2093-3371-4-7
|
| [40] |
Glaeser RM (2018) Proteins, interfaces, and cryo-EM grids. Curr Opin Colloid Interface Sci 34: 1-8. https://doi.org/10.1016/j.cocis.2017.12.009
|
| [41] |
D'Imprima E, Floris D, Joppe M, et al. (2019) Protein denaturation at the air-water interface and how to prevent it. Elife 8: e42747. https://doi.org/10.7554/eLife.42747.001
|
| [42] |
Pantelic RS, Suk JW, Magnuson CW, et al. (2011) Graphene: substrate preparation and introduction. J Struct Biol 174: 234-238. https://doi.org/10.1016/j.jsb.2010.10.002
|
| [43] |
Russo CJ, Passmore LA (2014) Controlling protein adsorption on graphene for cryo-EM using low-energy hydrogen plasmas. Nat Methods 11: 649-652. https://doi.org/10.1038/nmeth.2931
|
| [44] |
Russo CJ, Passmore LA (2014) Ultrastable gold substrates for electron cryomicroscopy. Science 346: 1377-1380. https://doi.org/10.1126/science.1259530
|
| [45] |
Glaeser RM (1979) Prospects for extending the resolution limit of the electron microscope. J Microsc 117: 77-91. https://doi.org/10.1111/j.1365-2818.1979.tb00232.x
|
| [46] |
Williams DB, Carter CB Transmission Electron Microscopy: A Textbook for Materials Science (2009).
|
| [47] |
Frank J (2006) Three-dimensional electron microscopy of macromolecular assemblies: visualization of biological molecules in their native state. Oxford University Press.
|
| [48] |
Williams DB, Carter CB (2009) Planar defects. Transmission Electron Microscopy . Boston: Springer 419-439. https://doi.org/10.1007/978-0-387-76501-3_25
|
| [49] |
Williams DB, Carter CB (2009) Phase-contrast images. Transmission Electron Microscopy : 389-405. https://doi.org/10.1007/978-0-387-76501-3_23
|
| [50] |
Wade RH (1992) A brief look at imaging and contrast transfer. Ultramicroscopy 46: 145-156. https://doi.org/10.1016/0304-3991(92)90011-8
|
| [51] | Kohl H, Reimer L (2008) Transmission Electron Microscopy: Physics of Image Formation. New York: Springer-Verlag. https://doi.org/10.1007/978-0-387-40093-8 |
| [52] |
Saad A, Ludtke SJ, Jakana J, et al. (2001) Fourier amplitude decay of electron cryomicroscopic images of single particles and effects on structure determination. J Struct Biol 133: 32-42. https://doi.org/10.1006/jsbi.2001.4330
|
| [53] |
Faruqi AR, Subramaniam S (2000) CCD detectors in high-resolution biological electron microscopy. Q Rev Biophys 33: 1-27. https://doi.org/10.1017/S0033583500003577
|
| [54] | Meyer R, Kirkland A Direct electron detector (2007). |
| [55] |
Li X, Mooney P, Zheng S, et al. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10: 584-590. https://doi.org/10.1038/nmeth.2472
|
| [56] |
Scheres SHW (2014) Beam-induced motion correction for sub-megadalton cryo-EM particles. Elife 3: e03665. https://doi.org/10.7554/eLife.03665.001
|
| [57] |
Shigematsu H, Sigworth FJ (2013) Noise models and cryo-EM drift correction with a direct-electron camera. Ultramicroscopy 131: 61-69. https://doi.org/10.1016/j.ultramic.2013.04.001
|
| [58] |
Nogales E (2016) The development of cryo-EM into a mainstream structural biology technique. Nat Methods 13: 24-27. https://doi.org/10.1038/nmeth.3694
|
| [59] |
McMullan G, Faruqi AR (2008) Electron microscope imaging of single particles using the Medipix2 detector. Nucl Instruments Methods Phys Res Sect A Accel Spectrometers, Detect Assoc Equip 591: 129-133. https://doi.org/10.1016/j.nima.2008.03.041
|
| [60] |
Brilot AF, Chen JZ, Cheng A, et al. (2012) Beam-induced motion of vitrified specimen on holey carbon film. J Struct Biol 177: 630-637. https://doi.org/10.1016/j.jsb.2012.02.003
|
| [61] |
Mindell JA, Grigorieff N (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142: 334-347. https://doi.org/10.1016/S1047-8477(03)00069-8
|
| [62] |
Rohou A, Grigorieff N (2015) CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216-221. https://doi.org/10.1016/j.jsb.2015.08.008
|
| [63] |
Zhang K (2016) Gctf: Real-time CTF determination and correction. J Struct Biol 193: 1-12. https://doi.org/10.1016/j.jsb.2015.11.003
|
| [64] | MacKay DJC (2003) Information Theory, Inference and Learning Algorithms. Cambridge university press. |
| [65] |
Sigworth FJ, Doerschuk PC, Carazo JM, et al. (2010) An introduction to maximum-likelihood methods in cryo-EM. Methods Enzym 482: 263-294. https://doi.org/10.1016/S0076-6879(10)82011-7
|
| [66] | Zarzecka U, Grinzato A, Kandiah E, et al. Functional analysis and cryo-electron microscopy of Campylobacter jejuni serine protease HtrA (2020). https://doi.org/10.1080/19490976.2020.1810532 |
| [67] |
Orlova EV, Saibil HR (2011) Structural analysis of macromolecular assemblies by electron microscopy. Chem Rev 111: 7710-7748. https://doi.org/10.1021/cr100353t
|
| [68] |
Scheres SHW (2012) A Bayesian view on cryo-EM structure determination. J Mol Biol 415: 406-418. https://doi.org/10.1016/j.jmb.2011.11.010
|
| [69] |
Liao HY, Frank J (2010) Definition and estimation of resolution in single-particle reconstructions. Structure 18: 768-775. https://doi.org/10.1016/j.str.2010.05.008
|
| [70] |
Böttcher B, Wynne SA, Crowther RA (1997) Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 386: 88-91. https://doi.org/10.1038/386088a0
|
| [71] |
Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333: 721-745. https://doi.org/10.1016/j.jmb.2003.07.013
|
| [72] |
Henderson R, Sali A, Baker ML, et al. (2012) Outcome of the first electron microscopy validation task force meeting. Structure 20: 205-214.
|
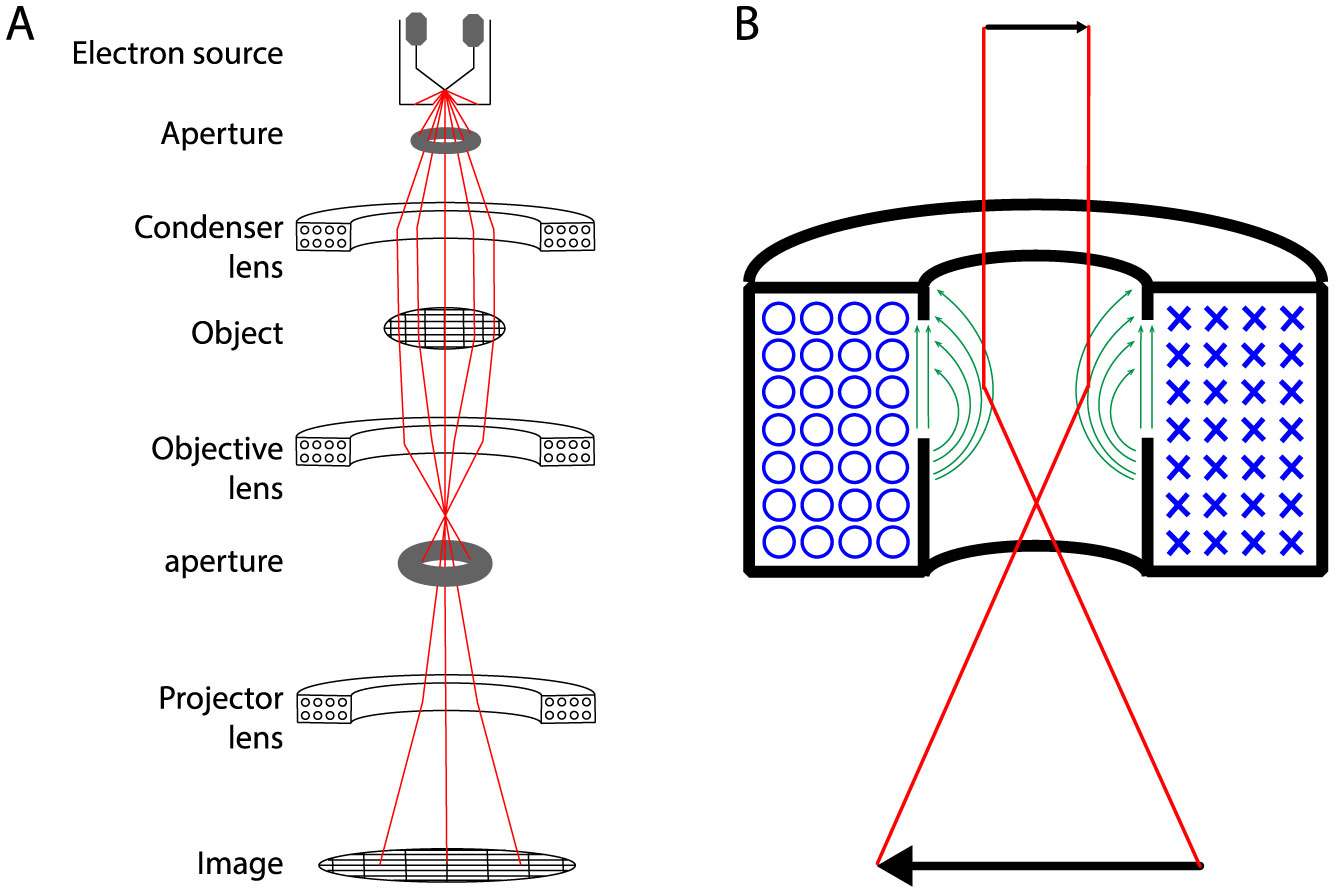
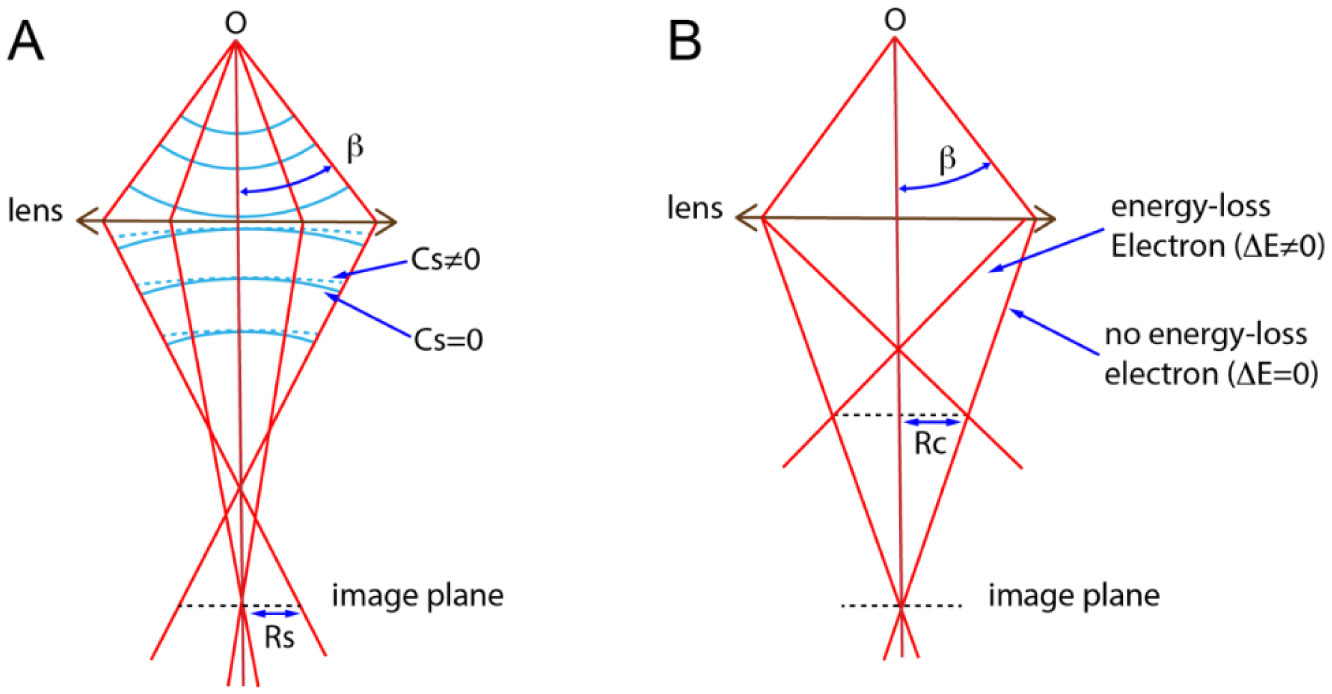
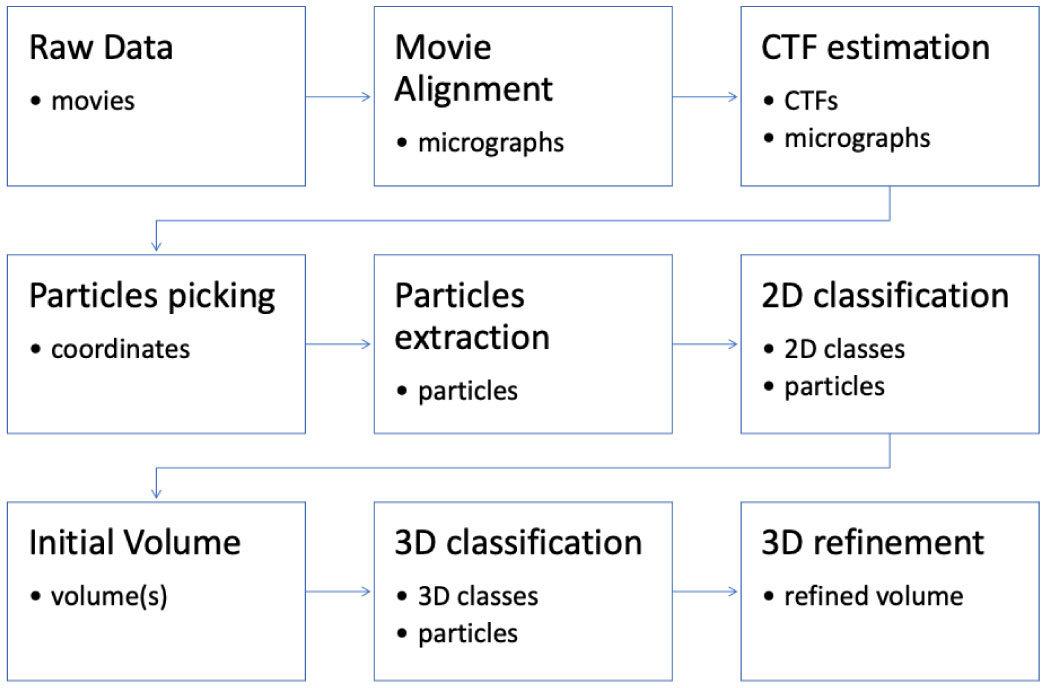
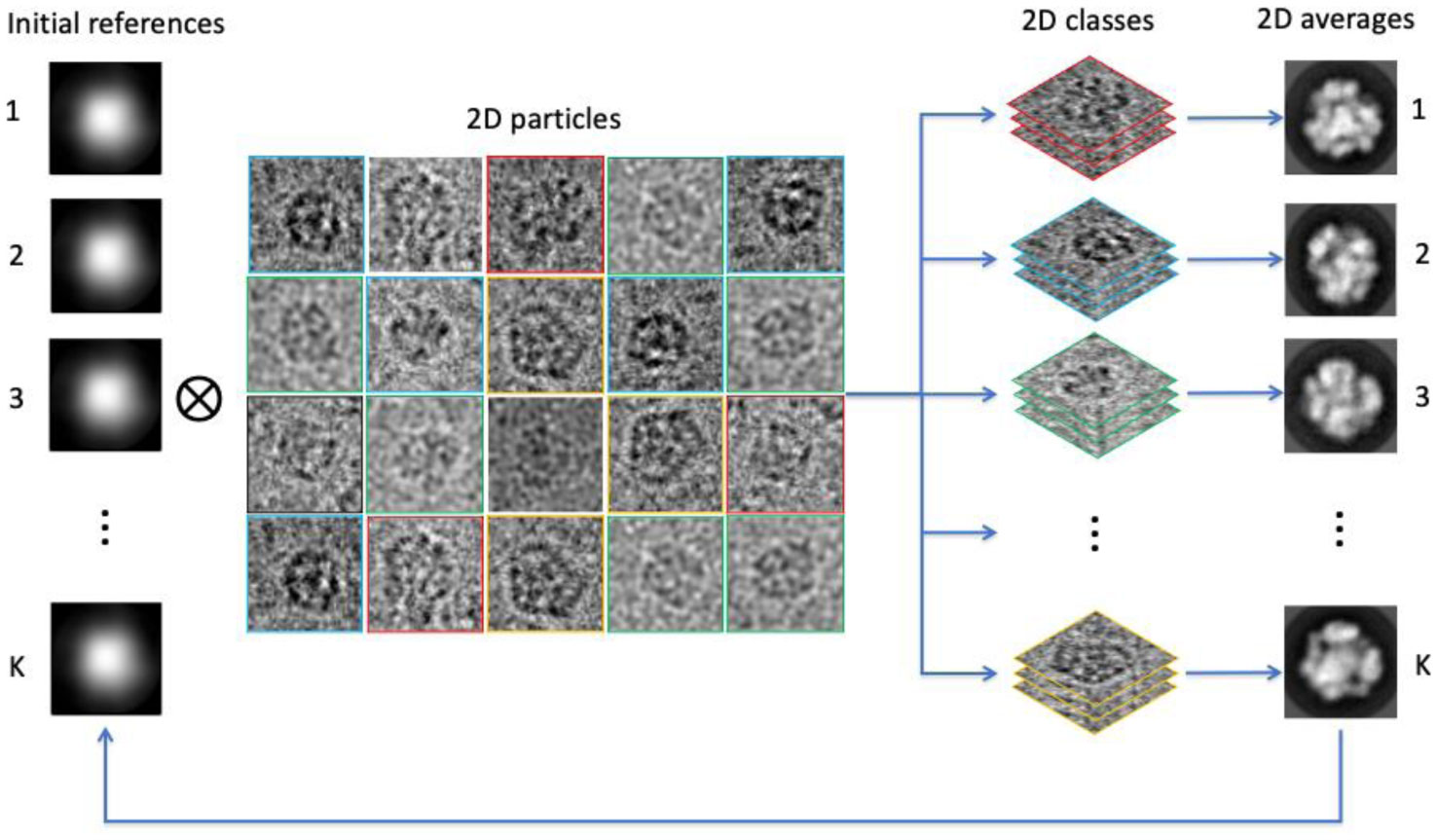

Figures(6)

Vittoria Raimondi, Alessandro Grinzato. A basic introduction to single particles cryo-electron microscopy[J]. AIMS Biophysics, 2022, 9(1): 5-20. doi: 10.3934/biophy.2022002







 DownLoad:
DownLoad: