Citation: Ateeq Al-Zahrani, Natasha Cant, Vassilis Kargas, Tracy Rimington, Luba Aleksandrov, John R. Riordan, Robert C. Ford. Structure of the cystic fibrosis transmembrane conductance regulator in the inward-facing conformation revealed by single particle electron microscopy[J]. AIMS Biophysics, 2015, 2(2): 131-152. doi: 10.3934/biophy.2015.2.131

| [1] |
Cutting GR (2005) Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet 6: 237-260. doi: 10.1146/annurev.genom.6.080604.162254

|
| [2] |
Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8: 67-113. doi: 10.1146/annurev.cb.08.110192.000435
|
| [3] |
Riordan JR, Rommens JM, Kerem B, et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066-1073. doi: 10.1126/science.2475911
|
| [4] |
Gray MA, Winpenny JP, Verdon B, et al. (1995) Chloride channels and cystic fibrosis of the pancreas. Biosci Rep 15: 531-541. doi: 10.1007/BF01204355
|
| [5] | McCarty NA (2000) Permeation through the CFTR chloride channel. J Exp Biol 203: 1947-1962. |
| [6] |
Vankeerberghen A, Cuppens H, Cassiman JJ (2002) The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions. J Cyst Fibros 1: 13-29. doi: 10.1016/S1569-1993(01)00003-0
|
| [7] |
Riordan JR (2008) CFTR function and prospects for therapy. Annu Rev Biochem 77: 701-726. doi: 10.1146/annurev.biochem.75.103004.142532
|
| [8] |
Loo MA, Jensen TJ, Cui L, et al. (1998) Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J 17: 6879-6887. doi: 10.1093/emboj/17.23.6879
|
| [9] | Lukacs GL, Chang XB, Bear C, et al. (1993) The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem 268: 21592-21598. |
| [10] | Lukacs GL, Segal G, Kartner N, et al. (1997) Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J 328 ( Pt 2): 353-361. |
| [11] | O'Ryan L, Rimington T, Cant N, et al. (2012) Expression and purification of the cystic fibrosis transmembrane conductance regulator protein in Saccharomyces cerevisiae. J Vis Exp. |
| [12] |
Huang P, Liu Q, Scarborough GA (1998) Lysophosphatidylglycerol: a novel effective detergent for solubilizing and purifying the cystic fibrosis transmembrane conductance regulator. Anal Biochem 259: 89-97. doi: 10.1006/abio.1998.2633
|
| [13] |
Huang P, Stroffekova K, Cuppoletti J, et al. (1996) Functional expression of the cystic fibrosis transmembrane conductance regulator in yeast. Biochim Biophys Acta 1281: 80-90. doi: 10.1016/0005-2736(96)00032-6
|
| [14] |
Ketchum CJ, Rajendrakumar GV, Maloney PC (2004) Characterization of the adenosinetriphosphatase and transport activities of purified cystic fibrosis transmembrane conductance regulator. Biochemistry 43: 1045-1053. doi: 10.1021/bi035382a
|
| [15] |
Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, et al. (2004) An evaluation of detergents for NMR structural studies of membrane proteins. J Biomol NMR 28: 43-57. doi: 10.1023/B:JNMR.0000012875.80898.8f
|
| [16] |
Yang Z, Wang C, Zhou Q, et al. (2014) Membrane protein stability can be compromised by detergent interactions with the extramembranous soluble domains. Protein Sci Public Protein Soci 23: 769-789. doi: 10.1002/pro.2460
|
| [17] |
Matar-Merheb R, Rhimi M, Leydier A, et al. (2011) Structuring detergents for extracting and stabilizing functional membrane proteins. PloS one 6: e18036. doi: 10.1371/journal.pone.0018036
|
| [18] |
Galian C, Manon F, Dezi M, et al. (2011) Optimized purification of a heterodimeric ABC transporter in a highly stable form amenable to 2-D crystallization. PloS one 6: e19677. doi: 10.1371/journal.pone.0019677
|
| [19] | Vinothkumar KR, Henderson R (2010) Structures of membrane proteins. Q Rev Biophys 43: 65-158. |
| [20] |
Guggino WB (2004) The cystic fibrosis transmembrane regulator forms macromolecular complexes with PDZ domain scaffold proteins. Proc Am Thorac Soc 1: 28-32. doi: 10.1513/pats.2306011
|
| [21] |
Karthikeyan S, Leung T, Birrane G, et al. (2001) Crystal structure of the PDZ1 domain of human Na(+)/H(+) exchanger regulatory factor provides insights into the mechanism of carboxyl-terminal leucine recognition by class I PDZ domains. J Mol Biol 308: 963-973. doi: 10.1006/jmbi.2001.4634
|
| [22] |
Karthikeyan S, Leung T, Ladias JA (2002) Structural determinants of the Na+/H+ exchanger regulatory factor interaction with the beta 2 adrenergic and platelet-derived growth factor receptors. J Biol Chem 277: 18973-18978. doi: 10.1074/jbc.M201507200
|
| [23] |
Li C, Naren AP (2011) Analysis of CFTR interactome in the macromolecular complexes. Methods Mol Biol 741: 255-270. doi: 10.1007/978-1-61779-117-8_17
|
| [24] |
Li C, Roy K, Dandridge K, et al. (2004) Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J Biol Chem 279: 24673-24684. doi: 10.1074/jbc.M400688200
|
| [25] |
Wang S, Yue H, Derin RB, et al. (2000) Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell 103: 169-179. doi: 10.1016/S0092-8674(00)00096-9
|
| [26] | Bozoky Z, Krzeminski M, Muhandiram R, et al. (2013) Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc Natl Acad Sci U S A. |
| [27] |
Guerra L, Favia M, Fanelli T, et al. (2004) Stimulation of Xenopus P2Y1 receptor activates CFTR in A6 cells. Pflugers Arch 449: 66-75. doi: 10.1007/s00424-004-1293-2
|
| [28] | Bossard F, Robay A, Toumaniantz G, et al. (2007) NHE-RF1 protein rescues DeltaF508-CFTR function. Am J Physiol Lung Cell Mol Physiol 292: L1085-1094. |
| [29] | Zhang L, Aleksandrov LA, Riordan JR, et al. (2011) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim Biophys Acta 1808: 399-404. |
| [30] |
Zhang L, Aleksandrov LA, Zhao ZF, et al. (2009) Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J Struct Biol 167: 242-251. doi: 10.1016/j.jsb.2009.06.004
|
| [31] |
Rosenberg MF, O'Ryan LP, Hughes G, et al. (2011) The cystic fibrosis transmembrane conductance regulator (CFTR): three-dimensional structure and localization of a channel gate. J Biol Chem 286: 42647-42654. doi: 10.1074/jbc.M111.292268
|
| [32] |
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443: 180-185. doi: 10.1038/nature05155
|
| [33] |
Ward A, Reyes CL, Yu J, et al. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A 104: 19005-19010. doi: 10.1073/pnas.0709388104
|
| [34] | Hildebrandt E, Zhang Q, Cant N, et al. (2014) A survey of detergents for the purification of stable, active human cystic fibrosis transmembrane conductance regulator (CFTR). Biochim Biophys Acta 1838: 2825-2837. |
| [35] | Al-Zahrani A (2014) Structural biology of Cystic Fibrosis Transmembrane Conductance Regulator, an ATP-binding cassette protein of medical importance. PhD Thesis, University of Manchester. |
| [36] | Pollock N, Cant N, Rimington T, et al. (2014) Purification of the cystic fibrosis transmembrane conductance regulator protein expressed in Saccharomyces cerevisiae. J Vis Exp: JoVE. |
| [37] |
Chifflet S, Torriglia A, Chiesa R, et al. (1988) A method for the determination of inorganic-phosphate in the presence of labile organic phosphate and high-concentrations of protein- application to lens ATPases. Anal Biochem 168: 1-4. doi: 10.1016/0003-2697(88)90002-4
|
| [38] |
Rothnie A, Theron D, Soceneantu L, et al. (2001) The importance of cholesterol in maintenance of P-glycoprotein activity and its membrane perturbing influence. Eur Biophys J Biophys Lett 30: 430-442. doi: 10.1007/s002490100156
|
| [39] | Cant N, Pollock N, Rimington T, et al. (2014) Purification of the Cystic Fibrosis Transmembrane Conductance Regulator Protein Expressed in Saccharomyces cerevisiae. J Vis Exp 87. |
| [40] |
Schultz BD, Bridges RJ, Frizzell RA (1996) Lack of conventional ATPase properties in CFTR chloride channel gating. J Membr Biol 151: 63-75. doi: 10.1007/s002329900058
|
| [41] |
Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82-97. doi: 10.1006/jsbi.1999.4174
|
| [42] |
Zhang L, Aleksandrov LA, Riordan JR, et al. (2011) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim Biophys Acta 1808: 399-404. doi: 10.1016/j.bbamem.2010.08.012
|
| [43] | Zhang L, Aleksandrov LA, Zhao Z, et al. (2009) Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J Struct Biol. |
| [44] |
Henderson R, Sali A, Baker ML, et al. (2012) Outcome of the first electron microscopy validation task force meeting. Structure 20: 205-214. doi: 10.1016/j.str.2011.12.014
|
| [45] |
Srinivasan V, Pierik AJ, Lill R (2014) Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 343: 1137-1140. doi: 10.1126/science.1246729
|
| [46] |
Claude JB, Suhre K, Notredame C, et al. (2004) CaspR: a web server for automated molecular replacement using homology modelling. Nucleic Acids Res 32: W606-609. doi: 10.1093/nar/gkh400
|
| [47] | Eswar N, Webb B, Marti-Renom MA, et al. (2007) Comparative protein structure modeling using MODELLER. Current protocols in protein science/editorial board, John E Coligan Chapter 2: Unit 2 9. |
| [48] | Laskowski RA, Rullmannn JA, MacArthur MW, et al. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477-486. |
| [49] | Yang Z, Lasker K, Schneidman-Duhovny D, et al. (2012) UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol 179: 269-278. |
| [50] | Zhang L, Aleksandrov LA, Riordan JR, et al. (2010) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim Biophys Acta. |
| [51] |
Zhang L, Aleksandrov LA, Zhao Z, et al. (2009) Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J Struct Biol 167: 242-251. doi: 10.1016/j.jsb.2009.06.004
|
| [52] |
Eckford PD, Li C, Ramjeesingh M, et al. (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 287: 36639-36649. doi: 10.1074/jbc.M112.393637
|
| [53] |
Venerando A, Franchin C, Cant N, et al. (2013) Detection of phospho-sites generated by protein kinase CK2 in CFTR: mechanistic aspects of Thr1471 phosphorylation. PloS one 8: e74232. doi: 10.1371/journal.pone.0074232
|
| [54] |
Kogan I, Ramjeesingh M, Huan LJ, et al. (2001) Perturbation of the pore of the cystic fibrosis transmembrane conductance regulator (CFTR) inhibits its atpase activity. J Biol Chem 276: 11575-11581. doi: 10.1074/jbc.M010403200
|
| [55] |
Aleksandrov AA, Kota P, Cui L, et al. (2012) Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. J Mol Biol 419: 41-60. doi: 10.1016/j.jmb.2012.03.001
|
| [56] |
Alexandrov AI, Mileni M, Chien EY, et al. (2008) Microscale fluorescent thermal stability assay for membrane proteins. Structure 16: 351-359. doi: 10.1016/j.str.2008.02.004
|
| [57] | Lysko KA, Carlson R, Taverna R, et al. (1981) Protein involvement in structural transition of erythrocyte ghosts. Use of thermal gel analysis to detect protein aggregation. Biochemistry 20: 5570-5576. |
| [58] |
Soler G, Mattingly JR, Martinez-Carrion M (1984) Effects of heating on the ion-gating function and structural domains of the acetylcholine receptor. Biochemistry 23: 4630-4636. doi: 10.1021/bi00315a018
|
| [59] | Al Zahrani A (2014) PhD Thesis, University of Manchester. |
| [60] | Karthikeyan S, Leung T, Birrane G, et al. (2001) Crystal structure of the PDZ1 domain of human Na(+)/H(+) exchanger regulatory factor provides insights into the mechanism of carboxyl-terminal leucine recognition by class I PDZ domains. J Mol Biol 308: 963-973. |
| [61] | Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774-797. |
| [62] | Karthikeyan S, Leung T, Ladias JA (2002) Structural determinants of the Na+/H+ exchanger regulatory factor interaction with the beta 2 adrenergic and platelet-derived growth factor receptors. J Biol Chem 277: 18973-18978. |
| [63] | Karthikeyan S, Leung T, Ladias JA (2001) Structural basis of the Na+/H+ exchanger regulatory factor PDZ1 interaction with the carboxyl-terminal region of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 276: 19683-19686. |
| [64] | Zhang L, Aleksandrov LA, Zhao Z, et al. (2009) Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J Struct Biol 167: 242-251. |
| [65] |
Gadsby DC, Vergani P, Csanady L (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440: 477-483. doi: 10.1038/nature04712
|
| [66] |
Ma T, Thiagarajah JR, Yang H, et al. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 110: 1651-1658. doi: 10.1172/JCI0216112
|
| [67] | Verkman AS (1990) Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol 259: C375-388. |
| [68] |
Wellhauser L, Kim Chiaw P, Pasyk S, et al. (2009) A small-molecule modulator interacts directly with deltaPhe508-CFTR to modify its ATPase activity and conformational stability. Mol Pharmacol 75: 1430-1438. doi: 10.1124/mol.109.055608
|
| [69] | Laverty G, Anttila A, Carty J, et al. (2012) CFTR mediated chloride secretion in the avian renal proximal tubule. Comp Biochem Physiol A Mol Integr Physiol 161: 53-60. |
| [70] | Cole SP (2014) Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Ann Rev Pharmacol Toxicol 54: 95-117. |
| [71] |
Paumi CM, Chuk M, Snider J, et al. (2009) ABC transporters in Saccharomyces cerevisiae and their interactors: new technology advances the biology of the ABCC (MRP) subfamily. Microbiol Mol Biol Rev 73: 577-593. doi: 10.1128/MMBR.00020-09
|
| [72] |
Maher P (2005) The effects of stress and aging on glutathione metabolism. Ageing Res Rev 4: 288-314. doi: 10.1016/j.arr.2005.02.005
|
| [73] |
Kogan I, Ramjeesingh M, Li C, et al. (2003) CFTR directly mediates nucleotide-regulated glutathione flux. Embo J 22: 1981-1989. doi: 10.1093/emboj/cdg194
|
| [74] | Roum JH, Buhl R, McElvaney NG, et al. (1993) Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol 75: 2419-2424. |
| [75] | Gao L, Kim KJ, Yankaskas JR, et al. (1999) Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol 277: L113-118. |
| [76] | Marson FA, Bertuzzo CS, Ribeiro AF, et al. (2014) Polymorphisms in the glutathione pathway modulate cystic fibrosis severity: a cross-sectional study. BMC Med Genet 15: 27. |
| [77] | Rubera I, Duranton C, Melis N, et al. (2013) Role of CFTR in oxidative stress and suicidal death of renal cells during cisplatin-induced nephrotoxicity. Cell Death Disease 4: e817. |
| [78] |
Cui G, Song B, Turki HW, et al. (2012) Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflugers Arch: Eur J Physiol 463: 405-418. doi: 10.1007/s00424-011-1035-1
|
| [79] |
Bozoky Z, Krzeminski M, Muhandiram R, et al. (2013) Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc Natl Acad Sci U S A 110: E4427-4436. doi: 10.1073/pnas.1315104110
|
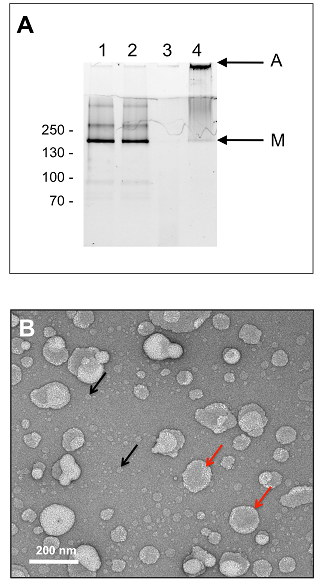
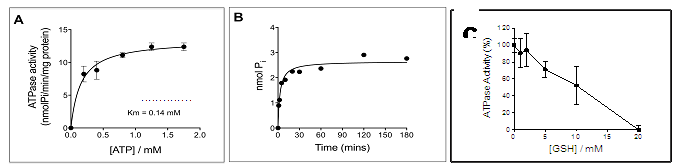
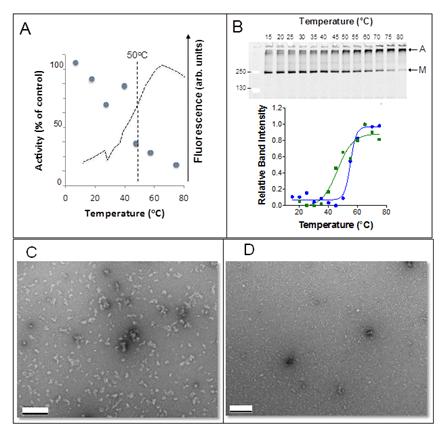
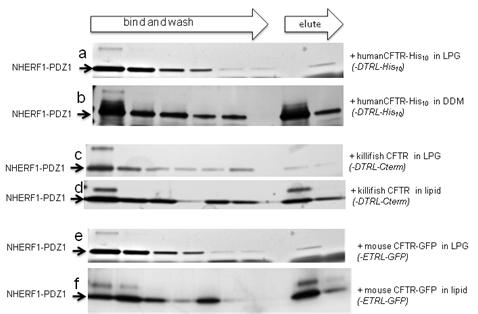
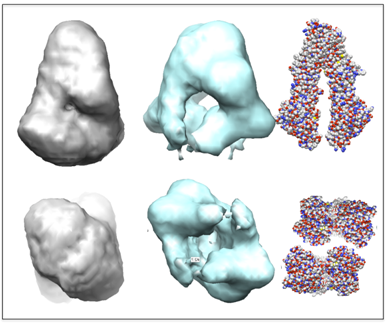
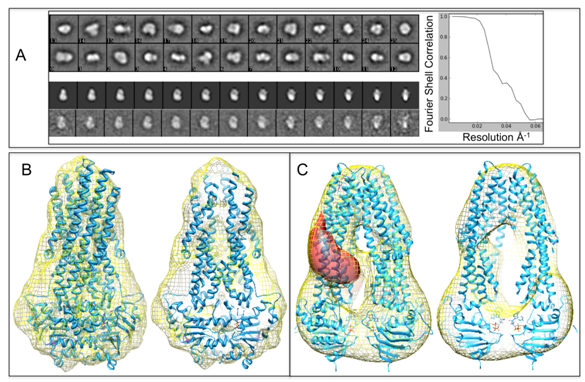
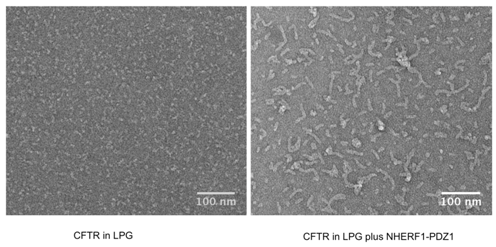
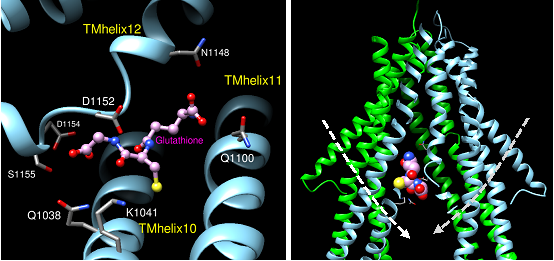
Figures(8) / Tables(1)

Ateeq Al-Zahrani, Natasha Cant, Vassilis Kargas, Tracy Rimington, Luba Aleksandrov, John R. Riordan, Robert C. Ford. Structure of the cystic fibrosis transmembrane conductance regulator in the inward-facing conformation revealed by single particle electron microscopy[J]. AIMS Biophysics, 2015, 2(2): 131-152. doi: 10.3934/biophy.2015.2.131







 DownLoad:
DownLoad: